User login
Solitary Plaque on the Nose
Solitary Plaque on the Nose
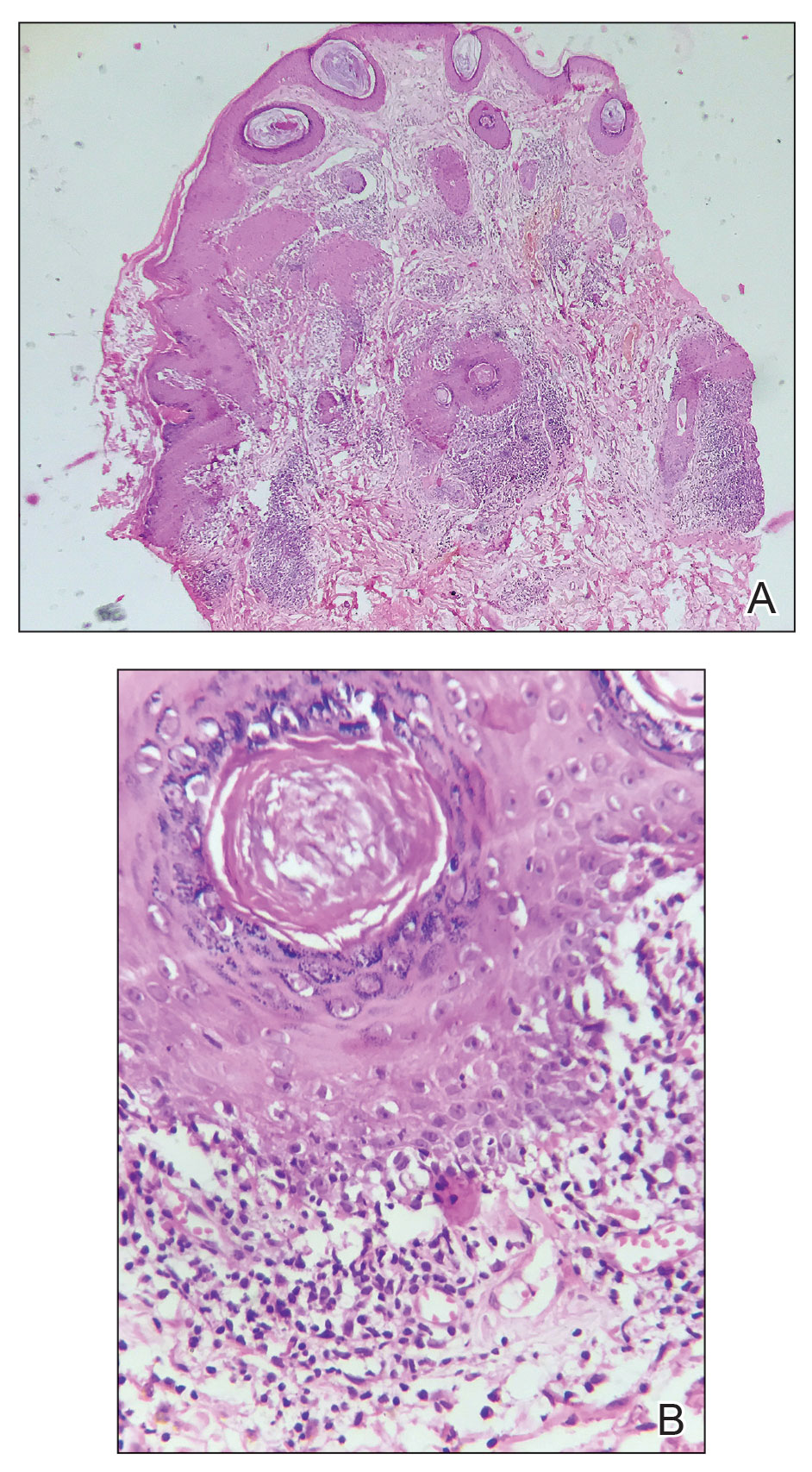
The biopsy revealed hyperkeratosis, hypergranulosis, follicular plugging, vacuolar interface dermatitis with apoptotic bodies, dyskeratotic keratinocytes, pigment incontinence, and melanophages. A perivascular, perifollicular, and periadnexal lymphoplasmacytic inflammatory infiltrate was noted in the superficial and deep dermis (Figure). Based on the characteristic clinical morphology, dermoscopic features, and histopathology, a diagnosis of discoid lupus erythematosus (DLE) was established. The patient was started on mometasone cream 0.1% and tacrolimus ointment 0.1% once daily, with strict recommendations for photoprotection. However, he subsequently was lost to follow-up, and treatment response could not be assessed.
Lupus erythematosus is a multisystemic autoimmune disease with a predilection for skin involvement that is characterized by the production of autoantibodies against nuclear antigens. Discoid lupus erythematosus is the predominant form of the disease, mostly affecting middle-aged women (female-to-male ratio, 4.1:1).1 Discoid lupus erythematosus usually manifests as well-demarcated, erythematous patches or plaques with partially adherent scales that extend into a patulous follicle. On removal, the scales show horny plugs underneath. This classic finding is known as the carpet tack sign.
As the lesions evolve, they expand with hyperpigmentation at the periphery as well as hypopigmentation, atrophy, scarring, and telangiectasias at the center.2 In our patient, the history of discharge and crusting of the lesion and the presence of slight central atrophy—all of which could be attributed to chronic application of topical medications such as corticosteroids, which can cause epidermal thinning, maceration, and secondary crust formation—raised clinical suspicion of cutaneous infections (eg, cutaneous leishmaniasis, lupus vulgaris) and squamous cell carcinoma. The presence of slightly raised margins upon clinical examination brought basal cell carcinoma (BCC) into the differential.
Dermoscopic features commonly seen in DLE reflect the pathologic findings. Follicular plugging and perifollicular white halos correspond to follicular hyperkeratosis and perifollicular fibrosis, respectively (eTable). Disease duration has been shown to alter the dermoscopic appearance of DLE with early active disease showing radially arranged arborizing blood vessels between perifollicular white halos along with follicular red dots, whereas lesions of longer duration display structureless white areas secondary to dermal fibrosis.3 Additionally, background erythema due to neoangiogenesis and dermal inflammation suggests that the disease is in its active state.

On dermoscopy, pigmentation structures such as brown dots, brown lines, and grey-brown dots and globules were seen more prominently in our patient with skin of color, making the underlying erythema more subtle than in patients with lighter skin types. Dotted and linear vessels also were seen in our patient, but not as prominently as typically is seen in lighter skin types.4
Lupus vulgaris was ruled out in our patient based on the absence of the typical orange to yellowish-orange background with vessels or any histopathologic evidence of epithelioid granulomas.5 Cutaneous leishmaniasis is characterized by polymorphic vascularization, erythema, follicular plugs, yellow-orange structureless areas with scales, and crusts on dermoscopy.6 Squamous cell carcinoma tends to show white structureless areas, looped vessels, and central keratin.7
Superficial BCC also appears as thin plaques or patches bound by a well-circumscribed, slightly raised, irregular margin. However, on dermoscopy, BCC typically exhibits spoke-wheel areas, arborizing vessels, comma vessels, and concentric structures.8
The clinical manifestations of crusting, discharge, and a raised border was atypical, probably owing to the long-term unsupervised application of topical medications, which made the initial diagnosis challenging. Therefore, various differential diagnoses were considered. Dermoscopic evaluation coupled with histology was performed, which ultimately confirmed the diagnosis of DLE.
- Gopalan G, Gopinath SR, Kothandaramasamy R, et al. A clinical and epidemiological study on discoid lupus erythematosus. Int J Res Dermatol 2018;4:396-402. doi:10.18203/issn.24554529.IntJRes Dermatol20183165
- McDaniel B, Sukumaran S, Koritala T, et al. Discoid lupus erythematosus. StatPearls [Internet]. StatPearls Publishing 2025. Updated August 28, 2023. Accessed October 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK493145/
- Fathy H, Ghanim BM, Refat S, et al. Dermoscopic criteria of discoid lupus erythematosus: an observational cross-sectional study of 28 patients. Indian J Dermatol Venereol Leprol 2022;88:360-366. doi:10.25259/IJDVL_207_19
- Ankad BS, Gupta A, Nikam BP, et al. Implications of dermoscopy and histopathological correlation in discoid lupus erythematosus in skin of color. Indian J Dermatol 2022;67:5‐11. doi:10.4103/ijd.ijd_591_21
- Jindal R, Chauhan P, Sethi S. Dermoscopy of the diverse spectrum of cutaneous tuberculosis in the skin of color. Dermatol Pract Concept. 2022;12:E2022203. doi:10.5826/dpc.1204a203
- Chauhan P, Adya KA. Dermatoscopy of cutaneous granulomatous disorders. Indian Dermatol Online J. 2021;12:34-44. doi:10.4103 /idoj.IDOJ_543_20.
- Rosendahl C, Cameron A, Argenziano G, et al. Dermoscopy of squamous cell carcinoma and keratoacanthoma. Arch Dermatol. 2012;148:1386-1392. doi:10.1001/archdermatol.2012.2974.
- Vinciullo C, Mada V. Basal cell carcinoma. 10th ed. Wiley: Blackwell Science; 2024.
The biopsy revealed hyperkeratosis, hypergranulosis, follicular plugging, vacuolar interface dermatitis with apoptotic bodies, dyskeratotic keratinocytes, pigment incontinence, and melanophages. A perivascular, perifollicular, and periadnexal lymphoplasmacytic inflammatory infiltrate was noted in the superficial and deep dermis (Figure). Based on the characteristic clinical morphology, dermoscopic features, and histopathology, a diagnosis of discoid lupus erythematosus (DLE) was established. The patient was started on mometasone cream 0.1% and tacrolimus ointment 0.1% once daily, with strict recommendations for photoprotection. However, he subsequently was lost to follow-up, and treatment response could not be assessed.
Lupus erythematosus is a multisystemic autoimmune disease with a predilection for skin involvement that is characterized by the production of autoantibodies against nuclear antigens. Discoid lupus erythematosus is the predominant form of the disease, mostly affecting middle-aged women (female-to-male ratio, 4.1:1).1 Discoid lupus erythematosus usually manifests as well-demarcated, erythematous patches or plaques with partially adherent scales that extend into a patulous follicle. On removal, the scales show horny plugs underneath. This classic finding is known as the carpet tack sign.
As the lesions evolve, they expand with hyperpigmentation at the periphery as well as hypopigmentation, atrophy, scarring, and telangiectasias at the center.2 In our patient, the history of discharge and crusting of the lesion and the presence of slight central atrophy—all of which could be attributed to chronic application of topical medications such as corticosteroids, which can cause epidermal thinning, maceration, and secondary crust formation—raised clinical suspicion of cutaneous infections (eg, cutaneous leishmaniasis, lupus vulgaris) and squamous cell carcinoma. The presence of slightly raised margins upon clinical examination brought basal cell carcinoma (BCC) into the differential.
Dermoscopic features commonly seen in DLE reflect the pathologic findings. Follicular plugging and perifollicular white halos correspond to follicular hyperkeratosis and perifollicular fibrosis, respectively (eTable). Disease duration has been shown to alter the dermoscopic appearance of DLE with early active disease showing radially arranged arborizing blood vessels between perifollicular white halos along with follicular red dots, whereas lesions of longer duration display structureless white areas secondary to dermal fibrosis.3 Additionally, background erythema due to neoangiogenesis and dermal inflammation suggests that the disease is in its active state.
On dermoscopy, pigmentation structures such as brown dots, brown lines, and grey-brown dots and globules were seen more prominently in our patient with skin of color, making the underlying erythema more subtle than in patients with lighter skin types. Dotted and linear vessels also were seen in our patient, but not as prominently as typically is seen in lighter skin types.4
Lupus vulgaris was ruled out in our patient based on the absence of the typical orange to yellowish-orange background with vessels or any histopathologic evidence of epithelioid granulomas.5 Cutaneous leishmaniasis is characterized by polymorphic vascularization, erythema, follicular plugs, yellow-orange structureless areas with scales, and crusts on dermoscopy.6 Squamous cell carcinoma tends to show white structureless areas, looped vessels, and central keratin.7
Superficial BCC also appears as thin plaques or patches bound by a well-circumscribed, slightly raised, irregular margin. However, on dermoscopy, BCC typically exhibits spoke-wheel areas, arborizing vessels, comma vessels, and concentric structures.8
The clinical manifestations of crusting, discharge, and a raised border was atypical, probably owing to the long-term unsupervised application of topical medications, which made the initial diagnosis challenging. Therefore, various differential diagnoses were considered. Dermoscopic evaluation coupled with histology was performed, which ultimately confirmed the diagnosis of DLE.
The biopsy revealed hyperkeratosis, hypergranulosis, follicular plugging, vacuolar interface dermatitis with apoptotic bodies, dyskeratotic keratinocytes, pigment incontinence, and melanophages. A perivascular, perifollicular, and periadnexal lymphoplasmacytic inflammatory infiltrate was noted in the superficial and deep dermis (Figure). Based on the characteristic clinical morphology, dermoscopic features, and histopathology, a diagnosis of discoid lupus erythematosus (DLE) was established. The patient was started on mometasone cream 0.1% and tacrolimus ointment 0.1% once daily, with strict recommendations for photoprotection. However, he subsequently was lost to follow-up, and treatment response could not be assessed.
Lupus erythematosus is a multisystemic autoimmune disease with a predilection for skin involvement that is characterized by the production of autoantibodies against nuclear antigens. Discoid lupus erythematosus is the predominant form of the disease, mostly affecting middle-aged women (female-to-male ratio, 4.1:1).1 Discoid lupus erythematosus usually manifests as well-demarcated, erythematous patches or plaques with partially adherent scales that extend into a patulous follicle. On removal, the scales show horny plugs underneath. This classic finding is known as the carpet tack sign.
As the lesions evolve, they expand with hyperpigmentation at the periphery as well as hypopigmentation, atrophy, scarring, and telangiectasias at the center.2 In our patient, the history of discharge and crusting of the lesion and the presence of slight central atrophy—all of which could be attributed to chronic application of topical medications such as corticosteroids, which can cause epidermal thinning, maceration, and secondary crust formation—raised clinical suspicion of cutaneous infections (eg, cutaneous leishmaniasis, lupus vulgaris) and squamous cell carcinoma. The presence of slightly raised margins upon clinical examination brought basal cell carcinoma (BCC) into the differential.
Dermoscopic features commonly seen in DLE reflect the pathologic findings. Follicular plugging and perifollicular white halos correspond to follicular hyperkeratosis and perifollicular fibrosis, respectively (eTable). Disease duration has been shown to alter the dermoscopic appearance of DLE with early active disease showing radially arranged arborizing blood vessels between perifollicular white halos along with follicular red dots, whereas lesions of longer duration display structureless white areas secondary to dermal fibrosis.3 Additionally, background erythema due to neoangiogenesis and dermal inflammation suggests that the disease is in its active state.
On dermoscopy, pigmentation structures such as brown dots, brown lines, and grey-brown dots and globules were seen more prominently in our patient with skin of color, making the underlying erythema more subtle than in patients with lighter skin types. Dotted and linear vessels also were seen in our patient, but not as prominently as typically is seen in lighter skin types.4
Lupus vulgaris was ruled out in our patient based on the absence of the typical orange to yellowish-orange background with vessels or any histopathologic evidence of epithelioid granulomas.5 Cutaneous leishmaniasis is characterized by polymorphic vascularization, erythema, follicular plugs, yellow-orange structureless areas with scales, and crusts on dermoscopy.6 Squamous cell carcinoma tends to show white structureless areas, looped vessels, and central keratin.7
Superficial BCC also appears as thin plaques or patches bound by a well-circumscribed, slightly raised, irregular margin. However, on dermoscopy, BCC typically exhibits spoke-wheel areas, arborizing vessels, comma vessels, and concentric structures.8
The clinical manifestations of crusting, discharge, and a raised border was atypical, probably owing to the long-term unsupervised application of topical medications, which made the initial diagnosis challenging. Therefore, various differential diagnoses were considered. Dermoscopic evaluation coupled with histology was performed, which ultimately confirmed the diagnosis of DLE.
- Gopalan G, Gopinath SR, Kothandaramasamy R, et al. A clinical and epidemiological study on discoid lupus erythematosus. Int J Res Dermatol 2018;4:396-402. doi:10.18203/issn.24554529.IntJRes Dermatol20183165
- McDaniel B, Sukumaran S, Koritala T, et al. Discoid lupus erythematosus. StatPearls [Internet]. StatPearls Publishing 2025. Updated August 28, 2023. Accessed October 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK493145/
- Fathy H, Ghanim BM, Refat S, et al. Dermoscopic criteria of discoid lupus erythematosus: an observational cross-sectional study of 28 patients. Indian J Dermatol Venereol Leprol 2022;88:360-366. doi:10.25259/IJDVL_207_19
- Ankad BS, Gupta A, Nikam BP, et al. Implications of dermoscopy and histopathological correlation in discoid lupus erythematosus in skin of color. Indian J Dermatol 2022;67:5‐11. doi:10.4103/ijd.ijd_591_21
- Jindal R, Chauhan P, Sethi S. Dermoscopy of the diverse spectrum of cutaneous tuberculosis in the skin of color. Dermatol Pract Concept. 2022;12:E2022203. doi:10.5826/dpc.1204a203
- Chauhan P, Adya KA. Dermatoscopy of cutaneous granulomatous disorders. Indian Dermatol Online J. 2021;12:34-44. doi:10.4103 /idoj.IDOJ_543_20.
- Rosendahl C, Cameron A, Argenziano G, et al. Dermoscopy of squamous cell carcinoma and keratoacanthoma. Arch Dermatol. 2012;148:1386-1392. doi:10.1001/archdermatol.2012.2974.
- Vinciullo C, Mada V. Basal cell carcinoma. 10th ed. Wiley: Blackwell Science; 2024.
- Gopalan G, Gopinath SR, Kothandaramasamy R, et al. A clinical and epidemiological study on discoid lupus erythematosus. Int J Res Dermatol 2018;4:396-402. doi:10.18203/issn.24554529.IntJRes Dermatol20183165
- McDaniel B, Sukumaran S, Koritala T, et al. Discoid lupus erythematosus. StatPearls [Internet]. StatPearls Publishing 2025. Updated August 28, 2023. Accessed October 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK493145/
- Fathy H, Ghanim BM, Refat S, et al. Dermoscopic criteria of discoid lupus erythematosus: an observational cross-sectional study of 28 patients. Indian J Dermatol Venereol Leprol 2022;88:360-366. doi:10.25259/IJDVL_207_19
- Ankad BS, Gupta A, Nikam BP, et al. Implications of dermoscopy and histopathological correlation in discoid lupus erythematosus in skin of color. Indian J Dermatol 2022;67:5‐11. doi:10.4103/ijd.ijd_591_21
- Jindal R, Chauhan P, Sethi S. Dermoscopy of the diverse spectrum of cutaneous tuberculosis in the skin of color. Dermatol Pract Concept. 2022;12:E2022203. doi:10.5826/dpc.1204a203
- Chauhan P, Adya KA. Dermatoscopy of cutaneous granulomatous disorders. Indian Dermatol Online J. 2021;12:34-44. doi:10.4103 /idoj.IDOJ_543_20.
- Rosendahl C, Cameron A, Argenziano G, et al. Dermoscopy of squamous cell carcinoma and keratoacanthoma. Arch Dermatol. 2012;148:1386-1392. doi:10.1001/archdermatol.2012.2974.
- Vinciullo C, Mada V. Basal cell carcinoma. 10th ed. Wiley: Blackwell Science; 2024.
Solitary Plaque on the Nose
Solitary Plaque on the Nose
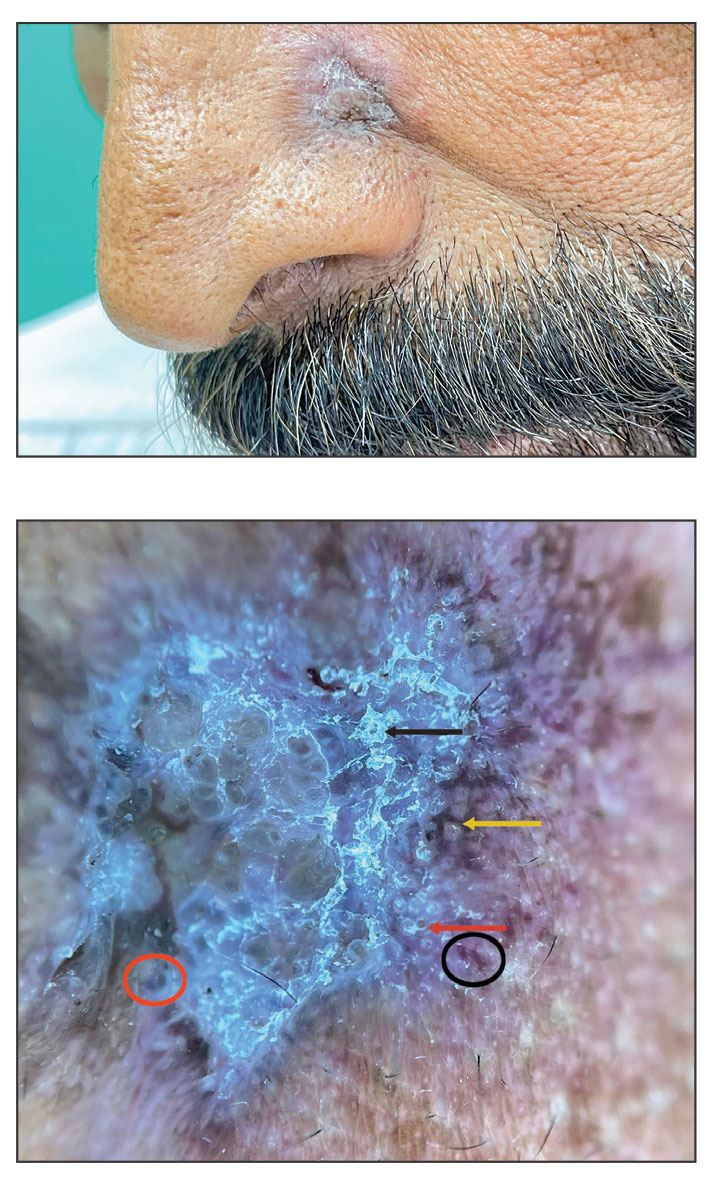
A 50-year-old Southeast Asian-Indian man presented to the dermatology clinic with a slightly elevated reddish-purple lesion on the left side of the nose accompanied by intense itching, occasional discharge, and crusting of 5 months’ duration. The patient reported applying multiple unknown topical agents initially prescribed to him by a physician; however, he subsequently continued applying these medications without regular follow-up visits. He had a history of smoking 2 packs per day for 25 years. His family history was unremarkable. Physical examination revealed a well-defined, 1.5×1.5-cm, nontender, scaly, erythematous to violaceous plaque with slightly raised margins, peripheral hyperpigmentation, and slight central atrophy on the left side of the nose. Dermoscopy revealed prominent follicles with a perifollicular halo (red arrow), white scales (black arrow), linear curved and dotted vessels (black circle), blue-grey globules (red circle), brown reticular lines (yellow arrow), and background erythema. General and systemic examination and routine laboratory workup were normal. A biopsy of the lesion was performed.

HPV Vaccine Reduces Immune Disease Risk in Women
TOPLINE: Human Papillomavirus (HPV) vaccination is associated with reduced risks of rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes among females aged 9 to 45 years. The analysis of 208,638 vaccinated individuals shows particularly strong protective effects in those aged 9 to 26 years and recipients of 9-valent HPV vaccines.
METHODOLOGY:
Researchers analyzed data from the US Collaborative Network in TriNetX spanning January 1, 2018, to December 20, 2022, enrolling 208,638 females aged 9 to 45 years who received HPV vaccination and matching them with 208,638 unvaccinated individuals using propensity scores.
Analysis included Cox proportional hazard regression to estimate hazard ratios and 95% CIs for immune-mediated diseases, with subgroup analyses stratified by age, race, smoking, obesity, asthma, and HPV vaccine types.
Participants were monitored from 31 days up to 365 days following their respective index dates, with sensitivity analyses conducted to evaluate short-term outcomes and compare results with influenza virus vaccine recipients.
TAKEAWAY:
HPV vaccination demonstrated reduced risks for rheumatoid arthritis (hazard ratio [HR], 0.487; 95% confidence interval [CI], 0.311-0.762), systemic lupus erythematosus (HR, 0.287; 95% CI, 0.179-0.460), and dermatomyositis (HR, 0.299; 95% CI, 0.098-0.908).
Recipients showed lower risks for inflammatory bowel disease (HR, 0.876; 95% CI, 0.811-0.946), celiac disease (HR, 0.400; 95% CI, 0.304-0.526), and type 1 diabetes (HR, 0.242; 95% CI, 0.184-0.318).
Subgroup analyses revealed significant risk reductions among females aged 9 to 26 years and those receiving 9-valent HPV vaccines compared to unvaccinated populations.
White and Black/African American individuals demonstrated reduced risks for various immune-mediated diseases, while Asians showed lower risks only for inflammatory bowel disease and overall immune-mediated diseases.
SOURCE: The study was led by Qianru Zhang, MD, Beijing Tsinghua Changgung Hospital in Beijing, China, James Cheng-Chung Wei, and Shiow-Ing Wang who contributed equally as first authors. It was published online in QJM: An International Journal of Medicine.
LIMITATIONS: According to the authors, research relying on Electronic Health Records (EHR) faced several constraints, including the absence of serial data on HPV antibody titers in vaccinated individuals and limited data regarding vaccination dosing numbers. Additionally, the current functionality of TriNetX prevented performing interaction terms in the statistical model for comprehensive subgroup analysis stratified by age, race, and vaccine types.
DISCLOSURES: The study received support from Chung Shan Medical University Hospital (Grant No. CSH-2023-E-001-Y2), Kaohsiung Veterans General Hospital (KSVGH 113-117), National Science and Technology Council (NSTC 112-2314-B-075B-020), and KSVNSU112-008. The funders had no role in the study's design, conduct, data analysis, or manuscript approval.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
TOPLINE: Human Papillomavirus (HPV) vaccination is associated with reduced risks of rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes among females aged 9 to 45 years. The analysis of 208,638 vaccinated individuals shows particularly strong protective effects in those aged 9 to 26 years and recipients of 9-valent HPV vaccines.
METHODOLOGY:
Researchers analyzed data from the US Collaborative Network in TriNetX spanning January 1, 2018, to December 20, 2022, enrolling 208,638 females aged 9 to 45 years who received HPV vaccination and matching them with 208,638 unvaccinated individuals using propensity scores.
Analysis included Cox proportional hazard regression to estimate hazard ratios and 95% CIs for immune-mediated diseases, with subgroup analyses stratified by age, race, smoking, obesity, asthma, and HPV vaccine types.
Participants were monitored from 31 days up to 365 days following their respective index dates, with sensitivity analyses conducted to evaluate short-term outcomes and compare results with influenza virus vaccine recipients.
TAKEAWAY:
HPV vaccination demonstrated reduced risks for rheumatoid arthritis (hazard ratio [HR], 0.487; 95% confidence interval [CI], 0.311-0.762), systemic lupus erythematosus (HR, 0.287; 95% CI, 0.179-0.460), and dermatomyositis (HR, 0.299; 95% CI, 0.098-0.908).
Recipients showed lower risks for inflammatory bowel disease (HR, 0.876; 95% CI, 0.811-0.946), celiac disease (HR, 0.400; 95% CI, 0.304-0.526), and type 1 diabetes (HR, 0.242; 95% CI, 0.184-0.318).
Subgroup analyses revealed significant risk reductions among females aged 9 to 26 years and those receiving 9-valent HPV vaccines compared to unvaccinated populations.
White and Black/African American individuals demonstrated reduced risks for various immune-mediated diseases, while Asians showed lower risks only for inflammatory bowel disease and overall immune-mediated diseases.
SOURCE: The study was led by Qianru Zhang, MD, Beijing Tsinghua Changgung Hospital in Beijing, China, James Cheng-Chung Wei, and Shiow-Ing Wang who contributed equally as first authors. It was published online in QJM: An International Journal of Medicine.
LIMITATIONS: According to the authors, research relying on Electronic Health Records (EHR) faced several constraints, including the absence of serial data on HPV antibody titers in vaccinated individuals and limited data regarding vaccination dosing numbers. Additionally, the current functionality of TriNetX prevented performing interaction terms in the statistical model for comprehensive subgroup analysis stratified by age, race, and vaccine types.
DISCLOSURES: The study received support from Chung Shan Medical University Hospital (Grant No. CSH-2023-E-001-Y2), Kaohsiung Veterans General Hospital (KSVGH 113-117), National Science and Technology Council (NSTC 112-2314-B-075B-020), and KSVNSU112-008. The funders had no role in the study's design, conduct, data analysis, or manuscript approval.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
TOPLINE: Human Papillomavirus (HPV) vaccination is associated with reduced risks of rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes among females aged 9 to 45 years. The analysis of 208,638 vaccinated individuals shows particularly strong protective effects in those aged 9 to 26 years and recipients of 9-valent HPV vaccines.
METHODOLOGY:
Researchers analyzed data from the US Collaborative Network in TriNetX spanning January 1, 2018, to December 20, 2022, enrolling 208,638 females aged 9 to 45 years who received HPV vaccination and matching them with 208,638 unvaccinated individuals using propensity scores.
Analysis included Cox proportional hazard regression to estimate hazard ratios and 95% CIs for immune-mediated diseases, with subgroup analyses stratified by age, race, smoking, obesity, asthma, and HPV vaccine types.
Participants were monitored from 31 days up to 365 days following their respective index dates, with sensitivity analyses conducted to evaluate short-term outcomes and compare results with influenza virus vaccine recipients.
TAKEAWAY:
HPV vaccination demonstrated reduced risks for rheumatoid arthritis (hazard ratio [HR], 0.487; 95% confidence interval [CI], 0.311-0.762), systemic lupus erythematosus (HR, 0.287; 95% CI, 0.179-0.460), and dermatomyositis (HR, 0.299; 95% CI, 0.098-0.908).
Recipients showed lower risks for inflammatory bowel disease (HR, 0.876; 95% CI, 0.811-0.946), celiac disease (HR, 0.400; 95% CI, 0.304-0.526), and type 1 diabetes (HR, 0.242; 95% CI, 0.184-0.318).
Subgroup analyses revealed significant risk reductions among females aged 9 to 26 years and those receiving 9-valent HPV vaccines compared to unvaccinated populations.
White and Black/African American individuals demonstrated reduced risks for various immune-mediated diseases, while Asians showed lower risks only for inflammatory bowel disease and overall immune-mediated diseases.
SOURCE: The study was led by Qianru Zhang, MD, Beijing Tsinghua Changgung Hospital in Beijing, China, James Cheng-Chung Wei, and Shiow-Ing Wang who contributed equally as first authors. It was published online in QJM: An International Journal of Medicine.
LIMITATIONS: According to the authors, research relying on Electronic Health Records (EHR) faced several constraints, including the absence of serial data on HPV antibody titers in vaccinated individuals and limited data regarding vaccination dosing numbers. Additionally, the current functionality of TriNetX prevented performing interaction terms in the statistical model for comprehensive subgroup analysis stratified by age, race, and vaccine types.
DISCLOSURES: The study received support from Chung Shan Medical University Hospital (Grant No. CSH-2023-E-001-Y2), Kaohsiung Veterans General Hospital (KSVGH 113-117), National Science and Technology Council (NSTC 112-2314-B-075B-020), and KSVNSU112-008. The funders had no role in the study's design, conduct, data analysis, or manuscript approval.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
Consider Cultural Practices and Barriers to Care When Treating Alopecia Areata
Consider Cultural Practices and Barriers to Care When Treating Alopecia Areata
The Comparison
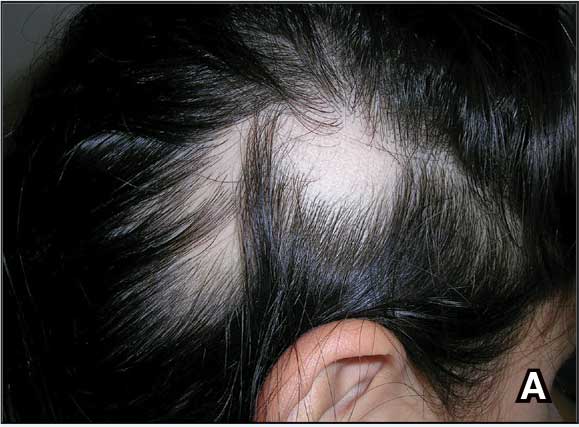
A. Alopecia areata in a young girl with a lighter skin tone. The fine white vellus hairs are signs of regrowth.
B. Alopecia areata in a 49-year-old man with tightly coiled hair and darker skin tone. Coiled white hairs are noted in the alopecia patches.
young girl with a lighter skin
tone. The fine white vellus
hairs are signs of regrowth. Photographs courtesy of
Richard P. Usatine, MD.

49-year-old man with tightly
coiled hair and darker skin
tone. Coiled white hairs
are noted in the alopecia
patches. Photographs courtesy of
Richard P. Usatine, MD.
Alopecia areata (AA) is a common autoimmune condition characterized by hair loss resulting from a T cell–mediated attack on the hair follicles. It manifests as nonscarring patches of hair loss on the scalp, eyebrows, eyelashes, and beard area as well as more extensive complete loss of scalp and body hair. While AA may affect individuals of any age, most patients develop their first patch(es) of hair loss during childhood.1 The treatment landscape for AA has evolved considerably in recent years, but barriers to access to newer treatments persist.
Epidemiology
AA is most prevalent among pediatric and adult individuals of African, Asian, or Hispanic/Latino descent.2-4 In some studies, Black individuals had higher odds and Asian individuals had lower odds of developing AA, while other studies have reported the highest standardized prevalence among Asian individuals.5 In the United States, AA affects about 1.47% of adults and as many as 0.11% of children.6-8 In Black patients, AA often manifests early with a female predominance.5
AA frequently is associated with autoimmune comorbidities, the most common being thyroid disease.3,5 In Black patients, AA is associated with more atopic comorbidities, including asthma, atopic dermatitis, and allergic rhinitis.5
Key Clinical Features
AA clinically manifests similarly across different skin tones; however, in patients with more tightly coiled or curly hair, the extent of scalp hair loss may be underestimated without a full examination. Culturally sensitive approaches to hair and scalp evaluation are essential, especially for Black women, whose hair care practices and scalp conditions may be overlooked or misunderstood during visits to evaluate hair loss. A thoughtful history and gentle examination of the hair and scalp that considers hair texture, cultural practices such as head coverings (eg, headwraps, turbans, hijabs), use of hair adornments (eg, clips, beads, bows), traditional braiding, and use of natural oils or herbal treatments, as well as styling methods including tight hairstyles, use of heat styling tools (eg, flat irons, curling irons), chemical application (eg, straighteners, hair color), and washing or styling frequency can improve diagnostic accuracy and help build trust in the patient-provider relationship.
Classic signs of AA visualized with dermoscopy include yellow and/or black dots on the scalp and exclamation point hairs. The appearance of fine white vellus hairs within the alopecic patches also may indicate early regrowth. On scalp trichoscopy, black dots are more prominent, and yellow dots are less prominent, in individuals with darker skin tones vs lighter skin tones.9
Worth Noting
In addition to a full examination of the scalp, documenting the extent of hair loss using validated severity scales, including the severity of alopecia tool (SALT), AA severity index (AASI), clinician-reported outcome assessment, and patient-reported outcome measures, can standardize disease severity assessment, facilitate timely insurance or medication approvals, and support objective tracking of treatment response, which may ultimately enhance access to care.10
Prompt treatment of AA is essential. Not surprisingly, patients given a diagnosis of AA may experience considerable emotional and psychological distress—regardless of the extent of the loss.11 Treatment options include mid- to high-potency topical or intralesional corticosteroids and newer and more targeted systemic options, including 3 Janus kinase (JAK) inhibitors—baricitinib, ritlecitinib, and deuruxolitinib—for more extensive disease.12 Treatment with intralesional corticosteroids may cause transient hypopigmentation, which may be more noticeable in patients with darker skin tones. Delays in treatment with JAK inhibitors can lead to a less-than-optimal response. Of the 3 JAK inhibitors that are approved by the US Food and Drug Administration for AA, only ritlecitinib is approved for children 12 years and older, leaving a therapeutic gap for younger patients that often leads to uncomfortable scalp injections, delayed or no treatment, off-label use of JAK inhibitors as well as the pairing of off-label dupilumab with oral minoxidil.12
Based on adult data, patients with severe disease and a shorter duration of hair loss (ie, < 4 years) tend to respond better to JAK inhibitors than those experiencing hair loss for longer periods. Also, those with more severe AA tend to have poorer outcomes than those with less severe disease.13 If treatment proves less than optimal, wigs and hair pieces may need to be considered. It is worth noting that some insurance companies will cover the cost of wigs for patients when prescribed as cranial prostheses.
Health Disparity Highlight
Health disparities in AA can be influenced by socioeconomic status and access to care. Patients from lower-income backgrounds often face barriers to accessing dermatologic care and treatments such as JAK inhibitors, which may remain inaccessible due to high costs and insurance limitations.14 These barriers can intersect with other factors such as age, sex, and race, potentially exacerbating disparities. Women with skin of color in underserved communities may experience delayed diagnosis, limited treatment options, and greater psychosocial distress from hair loss.14 Addressing these inequities requires advocacy, education for both patients and clinicians, and improved access to treatment to ensure comprehensive care for all patients.
- Kara T, Topkarcı Z. Interactions between posttraumatic stress disorder and alopecia areata in child with trauma exposure: two case reports. Int J Trichology. 2018;10:131-134. doi:10.4103/ijt.ijt_2_18
- Sy N, Mastacouris N, Strunk A, et al. Overall and racial and ethnic subgroup prevalences of alopecia areata, alopecia totalis, and alopecia universalis. JAMA Dermatol. 2023;159:419-423.
- Lee H, Jung SJ, Patel AB, et al. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol. 2020;83:1064-1070.
- Feaster B, McMichael AJ. Epidemiology of alopecia areata in Black patients: a retrospective chart review. J Am Acad Dermatol. 2022;87:1121-1123.
- Lee HH, Gwillim E, Patel KR, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:675-682.
- Mostaghimi A, Gao W, Ray M, et al. Trends in prevalence and incidence of alopecia areata, alopecia totalis, and alopecia universalis among adults and children in a US employer-sponsored insured population. JAMA Dermatol. 2023;159:411-418.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1(suppl 1):12-23.
- Karampinis E, Toli O, Georgopoulou KE, et al. Exploring pediatric dermatology in skin of color: focus on dermoscopy. Life (Basel). 2024;14:1604.
- King BA, Senna MM, Ohyama M, et al. Defining severity in alopecia areata: current perspectives and a multidimensional framework. Dermatol Ther (Heidelb). 2022;12:825-834.
- Toussi A, Barton VR, Le ST, et al. Psychosocial and psychiatric comorbidities and health-related quality of life in alopecia areata: a systematic review. J Am Acad Dermatol. 2021;85:162-175.
- Kalil L, Welch D, Heath CR, et al. Systemic therapies for pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1:36-42.
- King BA, Craiglow BG. Janus kinase inhibitors for alopecia areata. J Am Acad Dermatol. 2023;89:S29-S32.
- Klein EJ, Taiwò D, Kakpovbia E, et al. Disparities in Janus kinase inhibitor access for alopecia areata: a retrospective analysis. Int J Womens Dermatol. 2024;10:E155.
- McKenzie PL, Maltenfort M, Bruckner AL, et al. Evaluation of the prevalence and incidence of pediatric alopecia areata using electronic health record data. JAMA Dermatol. 2022;158:547-551. doi:10.1001/jamadermatol.2022.0351
The Comparison
A. Alopecia areata in a young girl with a lighter skin tone. The fine white vellus hairs are signs of regrowth.
B. Alopecia areata in a 49-year-old man with tightly coiled hair and darker skin tone. Coiled white hairs are noted in the alopecia patches.
young girl with a lighter skin
tone. The fine white vellus
hairs are signs of regrowth. Photographs courtesy of
Richard P. Usatine, MD.
49-year-old man with tightly
coiled hair and darker skin
tone. Coiled white hairs
are noted in the alopecia
patches. Photographs courtesy of
Richard P. Usatine, MD.
Alopecia areata (AA) is a common autoimmune condition characterized by hair loss resulting from a T cell–mediated attack on the hair follicles. It manifests as nonscarring patches of hair loss on the scalp, eyebrows, eyelashes, and beard area as well as more extensive complete loss of scalp and body hair. While AA may affect individuals of any age, most patients develop their first patch(es) of hair loss during childhood.1 The treatment landscape for AA has evolved considerably in recent years, but barriers to access to newer treatments persist.
Epidemiology
AA is most prevalent among pediatric and adult individuals of African, Asian, or Hispanic/Latino descent.2-4 In some studies, Black individuals had higher odds and Asian individuals had lower odds of developing AA, while other studies have reported the highest standardized prevalence among Asian individuals.5 In the United States, AA affects about 1.47% of adults and as many as 0.11% of children.6-8 In Black patients, AA often manifests early with a female predominance.5
AA frequently is associated with autoimmune comorbidities, the most common being thyroid disease.3,5 In Black patients, AA is associated with more atopic comorbidities, including asthma, atopic dermatitis, and allergic rhinitis.5
Key Clinical Features
AA clinically manifests similarly across different skin tones; however, in patients with more tightly coiled or curly hair, the extent of scalp hair loss may be underestimated without a full examination. Culturally sensitive approaches to hair and scalp evaluation are essential, especially for Black women, whose hair care practices and scalp conditions may be overlooked or misunderstood during visits to evaluate hair loss. A thoughtful history and gentle examination of the hair and scalp that considers hair texture, cultural practices such as head coverings (eg, headwraps, turbans, hijabs), use of hair adornments (eg, clips, beads, bows), traditional braiding, and use of natural oils or herbal treatments, as well as styling methods including tight hairstyles, use of heat styling tools (eg, flat irons, curling irons), chemical application (eg, straighteners, hair color), and washing or styling frequency can improve diagnostic accuracy and help build trust in the patient-provider relationship.
Classic signs of AA visualized with dermoscopy include yellow and/or black dots on the scalp and exclamation point hairs. The appearance of fine white vellus hairs within the alopecic patches also may indicate early regrowth. On scalp trichoscopy, black dots are more prominent, and yellow dots are less prominent, in individuals with darker skin tones vs lighter skin tones.9
Worth Noting
In addition to a full examination of the scalp, documenting the extent of hair loss using validated severity scales, including the severity of alopecia tool (SALT), AA severity index (AASI), clinician-reported outcome assessment, and patient-reported outcome measures, can standardize disease severity assessment, facilitate timely insurance or medication approvals, and support objective tracking of treatment response, which may ultimately enhance access to care.10
Prompt treatment of AA is essential. Not surprisingly, patients given a diagnosis of AA may experience considerable emotional and psychological distress—regardless of the extent of the loss.11 Treatment options include mid- to high-potency topical or intralesional corticosteroids and newer and more targeted systemic options, including 3 Janus kinase (JAK) inhibitors—baricitinib, ritlecitinib, and deuruxolitinib—for more extensive disease.12 Treatment with intralesional corticosteroids may cause transient hypopigmentation, which may be more noticeable in patients with darker skin tones. Delays in treatment with JAK inhibitors can lead to a less-than-optimal response. Of the 3 JAK inhibitors that are approved by the US Food and Drug Administration for AA, only ritlecitinib is approved for children 12 years and older, leaving a therapeutic gap for younger patients that often leads to uncomfortable scalp injections, delayed or no treatment, off-label use of JAK inhibitors as well as the pairing of off-label dupilumab with oral minoxidil.12
Based on adult data, patients with severe disease and a shorter duration of hair loss (ie, < 4 years) tend to respond better to JAK inhibitors than those experiencing hair loss for longer periods. Also, those with more severe AA tend to have poorer outcomes than those with less severe disease.13 If treatment proves less than optimal, wigs and hair pieces may need to be considered. It is worth noting that some insurance companies will cover the cost of wigs for patients when prescribed as cranial prostheses.
Health Disparity Highlight
Health disparities in AA can be influenced by socioeconomic status and access to care. Patients from lower-income backgrounds often face barriers to accessing dermatologic care and treatments such as JAK inhibitors, which may remain inaccessible due to high costs and insurance limitations.14 These barriers can intersect with other factors such as age, sex, and race, potentially exacerbating disparities. Women with skin of color in underserved communities may experience delayed diagnosis, limited treatment options, and greater psychosocial distress from hair loss.14 Addressing these inequities requires advocacy, education for both patients and clinicians, and improved access to treatment to ensure comprehensive care for all patients.
The Comparison
A. Alopecia areata in a young girl with a lighter skin tone. The fine white vellus hairs are signs of regrowth.
B. Alopecia areata in a 49-year-old man with tightly coiled hair and darker skin tone. Coiled white hairs are noted in the alopecia patches.
young girl with a lighter skin
tone. The fine white vellus
hairs are signs of regrowth. Photographs courtesy of
Richard P. Usatine, MD.
49-year-old man with tightly
coiled hair and darker skin
tone. Coiled white hairs
are noted in the alopecia
patches. Photographs courtesy of
Richard P. Usatine, MD.
Alopecia areata (AA) is a common autoimmune condition characterized by hair loss resulting from a T cell–mediated attack on the hair follicles. It manifests as nonscarring patches of hair loss on the scalp, eyebrows, eyelashes, and beard area as well as more extensive complete loss of scalp and body hair. While AA may affect individuals of any age, most patients develop their first patch(es) of hair loss during childhood.1 The treatment landscape for AA has evolved considerably in recent years, but barriers to access to newer treatments persist.
Epidemiology
AA is most prevalent among pediatric and adult individuals of African, Asian, or Hispanic/Latino descent.2-4 In some studies, Black individuals had higher odds and Asian individuals had lower odds of developing AA, while other studies have reported the highest standardized prevalence among Asian individuals.5 In the United States, AA affects about 1.47% of adults and as many as 0.11% of children.6-8 In Black patients, AA often manifests early with a female predominance.5
AA frequently is associated with autoimmune comorbidities, the most common being thyroid disease.3,5 In Black patients, AA is associated with more atopic comorbidities, including asthma, atopic dermatitis, and allergic rhinitis.5
Key Clinical Features
AA clinically manifests similarly across different skin tones; however, in patients with more tightly coiled or curly hair, the extent of scalp hair loss may be underestimated without a full examination. Culturally sensitive approaches to hair and scalp evaluation are essential, especially for Black women, whose hair care practices and scalp conditions may be overlooked or misunderstood during visits to evaluate hair loss. A thoughtful history and gentle examination of the hair and scalp that considers hair texture, cultural practices such as head coverings (eg, headwraps, turbans, hijabs), use of hair adornments (eg, clips, beads, bows), traditional braiding, and use of natural oils or herbal treatments, as well as styling methods including tight hairstyles, use of heat styling tools (eg, flat irons, curling irons), chemical application (eg, straighteners, hair color), and washing or styling frequency can improve diagnostic accuracy and help build trust in the patient-provider relationship.
Classic signs of AA visualized with dermoscopy include yellow and/or black dots on the scalp and exclamation point hairs. The appearance of fine white vellus hairs within the alopecic patches also may indicate early regrowth. On scalp trichoscopy, black dots are more prominent, and yellow dots are less prominent, in individuals with darker skin tones vs lighter skin tones.9
Worth Noting
In addition to a full examination of the scalp, documenting the extent of hair loss using validated severity scales, including the severity of alopecia tool (SALT), AA severity index (AASI), clinician-reported outcome assessment, and patient-reported outcome measures, can standardize disease severity assessment, facilitate timely insurance or medication approvals, and support objective tracking of treatment response, which may ultimately enhance access to care.10
Prompt treatment of AA is essential. Not surprisingly, patients given a diagnosis of AA may experience considerable emotional and psychological distress—regardless of the extent of the loss.11 Treatment options include mid- to high-potency topical or intralesional corticosteroids and newer and more targeted systemic options, including 3 Janus kinase (JAK) inhibitors—baricitinib, ritlecitinib, and deuruxolitinib—for more extensive disease.12 Treatment with intralesional corticosteroids may cause transient hypopigmentation, which may be more noticeable in patients with darker skin tones. Delays in treatment with JAK inhibitors can lead to a less-than-optimal response. Of the 3 JAK inhibitors that are approved by the US Food and Drug Administration for AA, only ritlecitinib is approved for children 12 years and older, leaving a therapeutic gap for younger patients that often leads to uncomfortable scalp injections, delayed or no treatment, off-label use of JAK inhibitors as well as the pairing of off-label dupilumab with oral minoxidil.12
Based on adult data, patients with severe disease and a shorter duration of hair loss (ie, < 4 years) tend to respond better to JAK inhibitors than those experiencing hair loss for longer periods. Also, those with more severe AA tend to have poorer outcomes than those with less severe disease.13 If treatment proves less than optimal, wigs and hair pieces may need to be considered. It is worth noting that some insurance companies will cover the cost of wigs for patients when prescribed as cranial prostheses.
Health Disparity Highlight
Health disparities in AA can be influenced by socioeconomic status and access to care. Patients from lower-income backgrounds often face barriers to accessing dermatologic care and treatments such as JAK inhibitors, which may remain inaccessible due to high costs and insurance limitations.14 These barriers can intersect with other factors such as age, sex, and race, potentially exacerbating disparities. Women with skin of color in underserved communities may experience delayed diagnosis, limited treatment options, and greater psychosocial distress from hair loss.14 Addressing these inequities requires advocacy, education for both patients and clinicians, and improved access to treatment to ensure comprehensive care for all patients.
- Kara T, Topkarcı Z. Interactions between posttraumatic stress disorder and alopecia areata in child with trauma exposure: two case reports. Int J Trichology. 2018;10:131-134. doi:10.4103/ijt.ijt_2_18
- Sy N, Mastacouris N, Strunk A, et al. Overall and racial and ethnic subgroup prevalences of alopecia areata, alopecia totalis, and alopecia universalis. JAMA Dermatol. 2023;159:419-423.
- Lee H, Jung SJ, Patel AB, et al. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol. 2020;83:1064-1070.
- Feaster B, McMichael AJ. Epidemiology of alopecia areata in Black patients: a retrospective chart review. J Am Acad Dermatol. 2022;87:1121-1123.
- Lee HH, Gwillim E, Patel KR, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:675-682.
- Mostaghimi A, Gao W, Ray M, et al. Trends in prevalence and incidence of alopecia areata, alopecia totalis, and alopecia universalis among adults and children in a US employer-sponsored insured population. JAMA Dermatol. 2023;159:411-418.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1(suppl 1):12-23.
- Karampinis E, Toli O, Georgopoulou KE, et al. Exploring pediatric dermatology in skin of color: focus on dermoscopy. Life (Basel). 2024;14:1604.
- King BA, Senna MM, Ohyama M, et al. Defining severity in alopecia areata: current perspectives and a multidimensional framework. Dermatol Ther (Heidelb). 2022;12:825-834.
- Toussi A, Barton VR, Le ST, et al. Psychosocial and psychiatric comorbidities and health-related quality of life in alopecia areata: a systematic review. J Am Acad Dermatol. 2021;85:162-175.
- Kalil L, Welch D, Heath CR, et al. Systemic therapies for pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1:36-42.
- King BA, Craiglow BG. Janus kinase inhibitors for alopecia areata. J Am Acad Dermatol. 2023;89:S29-S32.
- Klein EJ, Taiwò D, Kakpovbia E, et al. Disparities in Janus kinase inhibitor access for alopecia areata: a retrospective analysis. Int J Womens Dermatol. 2024;10:E155.
- McKenzie PL, Maltenfort M, Bruckner AL, et al. Evaluation of the prevalence and incidence of pediatric alopecia areata using electronic health record data. JAMA Dermatol. 2022;158:547-551. doi:10.1001/jamadermatol.2022.0351
- Kara T, Topkarcı Z. Interactions between posttraumatic stress disorder and alopecia areata in child with trauma exposure: two case reports. Int J Trichology. 2018;10:131-134. doi:10.4103/ijt.ijt_2_18
- Sy N, Mastacouris N, Strunk A, et al. Overall and racial and ethnic subgroup prevalences of alopecia areata, alopecia totalis, and alopecia universalis. JAMA Dermatol. 2023;159:419-423.
- Lee H, Jung SJ, Patel AB, et al. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol. 2020;83:1064-1070.
- Feaster B, McMichael AJ. Epidemiology of alopecia areata in Black patients: a retrospective chart review. J Am Acad Dermatol. 2022;87:1121-1123.
- Lee HH, Gwillim E, Patel KR, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:675-682.
- Mostaghimi A, Gao W, Ray M, et al. Trends in prevalence and incidence of alopecia areata, alopecia totalis, and alopecia universalis among adults and children in a US employer-sponsored insured population. JAMA Dermatol. 2023;159:411-418.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1(suppl 1):12-23.
- Karampinis E, Toli O, Georgopoulou KE, et al. Exploring pediatric dermatology in skin of color: focus on dermoscopy. Life (Basel). 2024;14:1604.
- King BA, Senna MM, Ohyama M, et al. Defining severity in alopecia areata: current perspectives and a multidimensional framework. Dermatol Ther (Heidelb). 2022;12:825-834.
- Toussi A, Barton VR, Le ST, et al. Psychosocial and psychiatric comorbidities and health-related quality of life in alopecia areata: a systematic review. J Am Acad Dermatol. 2021;85:162-175.
- Kalil L, Welch D, Heath CR, et al. Systemic therapies for pediatric alopecia areata. Pediatr Dermatol. 2025;42 suppl 1:36-42.
- King BA, Craiglow BG. Janus kinase inhibitors for alopecia areata. J Am Acad Dermatol. 2023;89:S29-S32.
- Klein EJ, Taiwò D, Kakpovbia E, et al. Disparities in Janus kinase inhibitor access for alopecia areata: a retrospective analysis. Int J Womens Dermatol. 2024;10:E155.
- McKenzie PL, Maltenfort M, Bruckner AL, et al. Evaluation of the prevalence and incidence of pediatric alopecia areata using electronic health record data. JAMA Dermatol. 2022;158:547-551. doi:10.1001/jamadermatol.2022.0351
Consider Cultural Practices and Barriers to Care When Treating Alopecia Areata
Consider Cultural Practices and Barriers to Care When Treating Alopecia Areata

A Systemic Lupus Erythematosus Incidence Surveillance Report Among DoD Beneficiaries During the COVID-19 Pandemic
A Systemic Lupus Erythematosus Incidence Surveillance Report Among DoD Beneficiaries During the COVID-19 Pandemic
Systemic lupus erythematosus (SLE), or lupus, is a rare autoimmune disease estimated to occur in about 5.1 cases per 100,000 person-years in the United States in 2018.1 The disease predominantly affects females, with an incidence of 8.7 cases per 100,000 person-years vs 1.2 cases per 100,000 person-years in males, and is most common in patients aged 15 to 44 years.1,2
Lupus presents with a constellation of clinical signs and symptoms that evolve, along with hallmark laboratory findings indicative of immune dysregulation and polyclonal B-cell activation. Consequently, a wide array of autoantibodies may be produced, although the combination of epitope specificity can vary from patient to patient.3 Nevertheless, > 98% of individuals diagnosed with lupus produce antinuclear antibodies (ANA), making ANA positivity a near-universal serologic feature at the time of diagnosis.
The pathogenesis of lupus is complex. Research from the past 5 decades supports the role of certain viral infections—such as Epstein-Barr virus (EBV) and cytomegalovirus—as potential triggers.4 These viruses are thought to initiate disease through mechanisms including activation of interferon pathways, exposure of cryptic intracellular antigens, molecular mimicry, and epitope spreading. Subsequent clonal expansion and autoantibody production occur to varying degrees, influenced by viral load and host susceptibility factors.
During the COVID-19 pandemic, it became evident that SARS-CoV-2 exerts profound effects on immune regulation, influencing infection outcomes through mechanisms such as hyperactivation of innate immunity, especially in the lungs, leading to acute respiratory distress syndrome. Additionally, SARS-CoV-2 has been associated with polyclonal B-cell activation and the generation of autoantibodies. This association gained attention after Bastard et al identified anti–type I interferon antibodies in patients with severe COVID-19, predominantly among males with a genetic predisposition. These autoantibodies were shown to impair antiviral defenses and contribute to life-threatening pneumonia.5
Subsequent studies demonstrated the production of a wide spectrum of functional autoantibodies, including ANA, in patients with COVID-19.6,7 These findings were attributed to the acute expansion of autoreactive clones among naïve-derived immunoglobulin G1 antibody-secreting cells during the early stages of infection, with the degree of expansion correlating with disease severity.8,9 Although longitudinal data up to 15 months postinfection suggest this serologic abnormality resolves in more than two-thirds of patients, the number of individuals infected globally has raised serious public health concerns regarding the potential long-term sequelae, including the onset of lupus or other autoimmune diseases in COVID-19 survivors.6-9 A limited number of case reports describing the onset of lupus following SARS-CoV-2 infection support this hypothesis.10
This surveillance analysis investigates lupus incidence among patients within the Military Health System (MHS), encompassing all TRICARE beneficiaries, from January 2018 to December 2022. The objective of this analysis was to examine lupus incidence trends throughout the COVID-19 pandemic, stratified by sex, age, and active-duty status.
Methods
The MHS provides health care services to about 9.5 million US Department of Defense (DoD) beneficiaries. Outpatient health records and laboratory results for individuals receiving care at military treatment facilities (MTFs) between January 1, 2018, and December 31, 2022, were obtained from the Comprehensive Ambulatory/ Professional Encounter Record and MHS GENESIS. For beneficiaries receiving care in the private sector, data were sourced from the TRICARE Encounter Data—Non-Institutional database.
Laboratory test results, including ANA testing, were available only for individuals receiving care at MTFs. These laboratory data were extracted from the Composite Health Care System Chemistry database and MHS GENESIS laboratory systems for the same time frame. Inpatient data were not included in this analysis. Data from 2017 were used solely as a look-back (or washout) period to identify and exclude prevalent lupus cases diagnosed before 2018 and were not included in the final results.
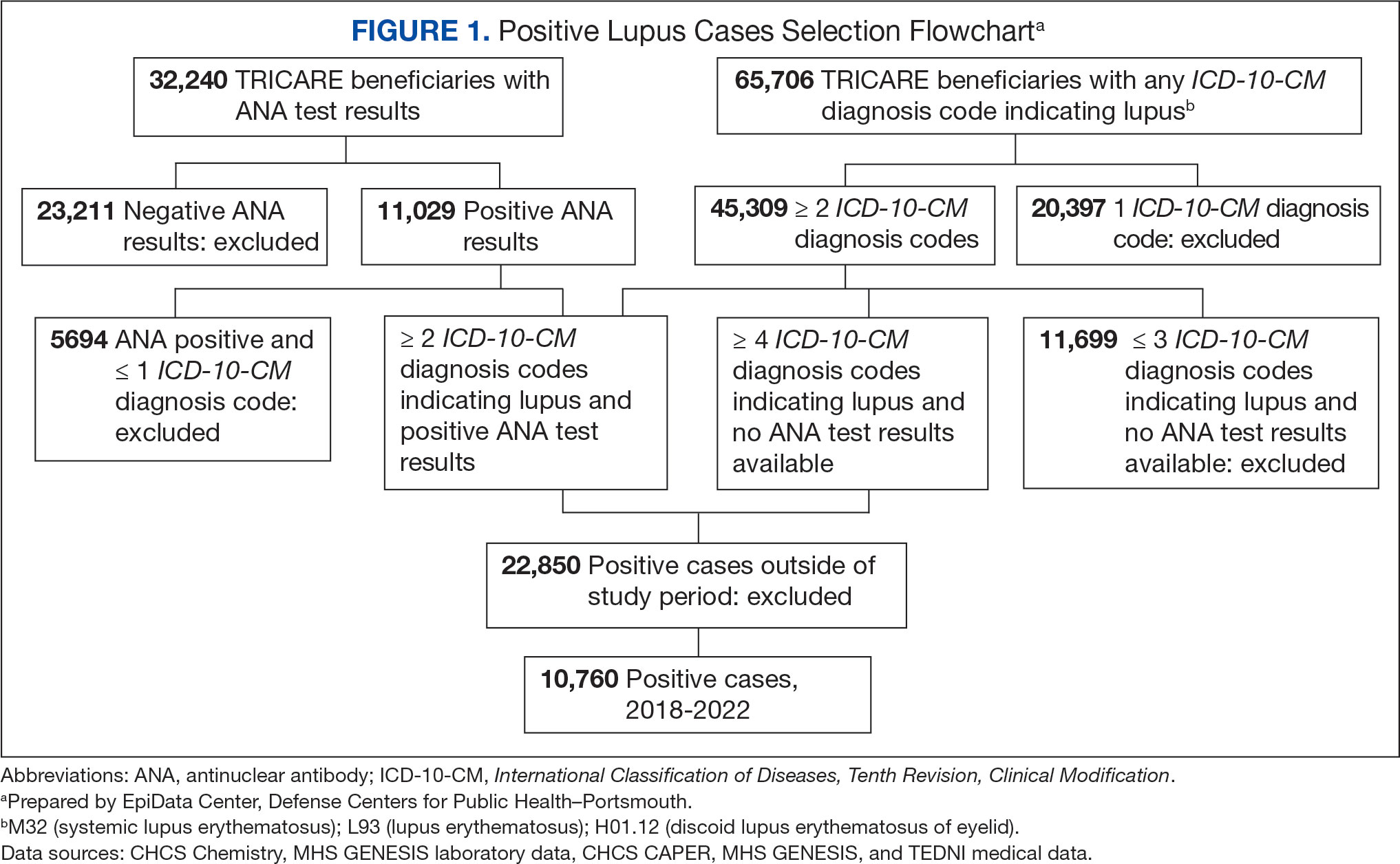
Lupus cases were identified by the presence of a positive ANA test and appropriate International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) codes. A positive ANA result was defined as either a qualitative result marked positive or a titer ≥ 1:80. The ICD-10-CM codes considered indicative of lupus included variations of M32, L93, or H01.12.
M32, L93, or H01.12. For cases with a positive ANA test, a lupus diagnosis required the presence of ≥ 2 lupus related ICD-10-CM codes. In the absence of ANA test results, a stricter criterion was applied: ≥ 4 lupus ICD-10-CM diagnosis codes recorded on separate days were required for inclusion.
Beneficiaries were excluded if they had a negative ANA result, only 1 lupus ICD- 10-CM diagnosis code, 1 positive ANA with only 1 corresponding ICD-10-CM code, or if their diagnosis occurred outside the defined study period. Patients and members of the public were not involved in the design, conduct, reporting, or dissemination of this study.
Results
Between January 1, 2017, and December 31, 2022, 99,946 TRICARE beneficiaries had some indication of lupus testing or diagnosis in their health records (Figure 1). Of these beneficiaries, 5335 had a positive ANA result and ≥ 2 ICD-10-CM lupus diagnosis codes. An additional 28,275 beneficiaries had ≥ 4 ICD-10-CM lupus diagnosis codes but no ANA test results. From these groups, the final sample included 10,760 beneficiaries who met the incident case definitions for SLE during the study period (2018 through 2022).
Most cases (85.1%, n = 9157) were diagnosed through TRICARE claims, while 1205 (11.2%) were diagnosed within the MHS. Another 398 (3.7%) had documentation of care both within and outside the MHS. Incident SLE cases declined by an average of 16% annually during the study period (Figure 2). This trend amounted to an overall reduction of 48.2%, from 2866 cases in 2018 to 1399 cases in 2022. This decline occurred despite total medical encounters among DoD beneficiaries remaining relatively stable during the pandemic years, with only a 3.5% change between 2018 and 2022.
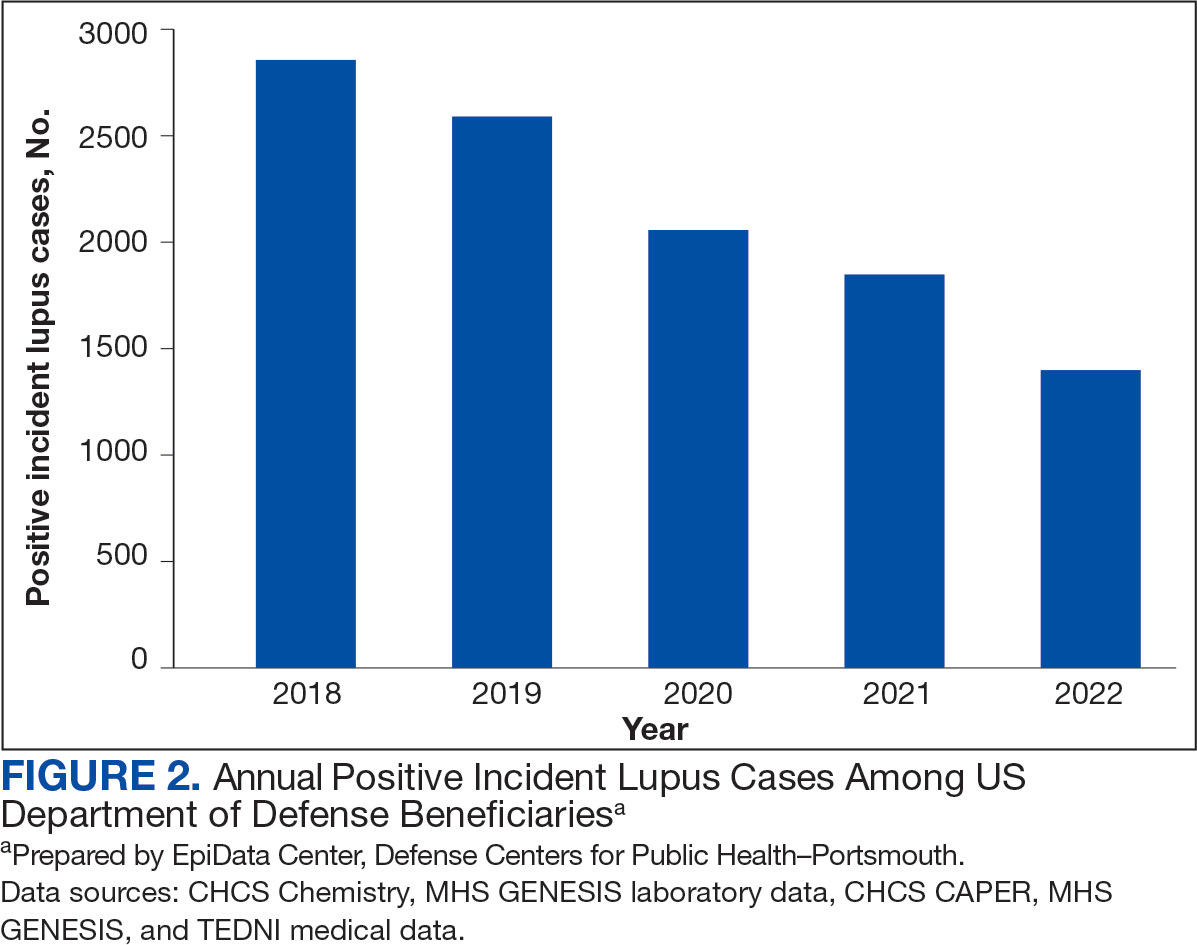
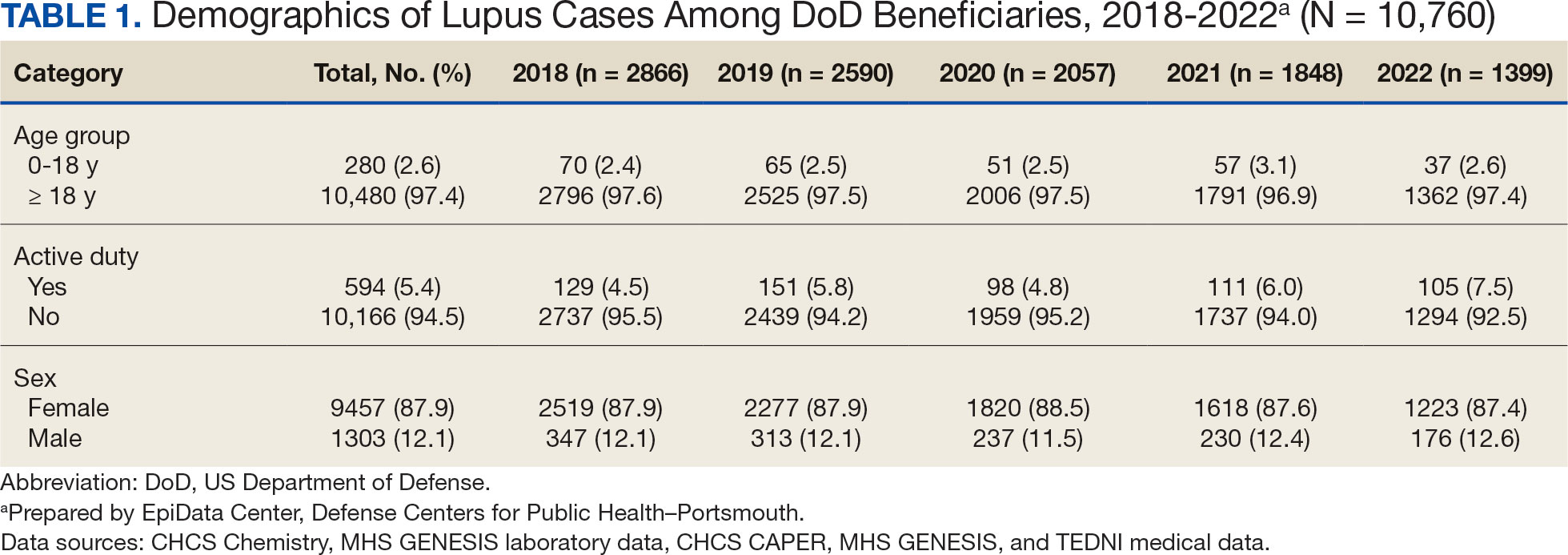
The disease was more prevalent among female beneficiaries, with a female to- male ratio of 7:1 (Table 1). Among women, the number of new cases declined from 2519 in 2018 to 1223 in 2022, while the number of cases among men remained consistently < 350 annually. Similar trends were observed across other strata. Incident SLE cases were more common among nonactive-duty beneficiaries than active-duty service members, with a ratio of 18:1. New cases among active-duty members remained < 155 per year. Age-stratified data revealed that SLE was diagnosed predominantly in individuals aged ≥ 18 years, with a ratio of 37:1 compared with individuals aged < 18 years. Among children, the number of new cases remained < 75 per year throughout the study period.
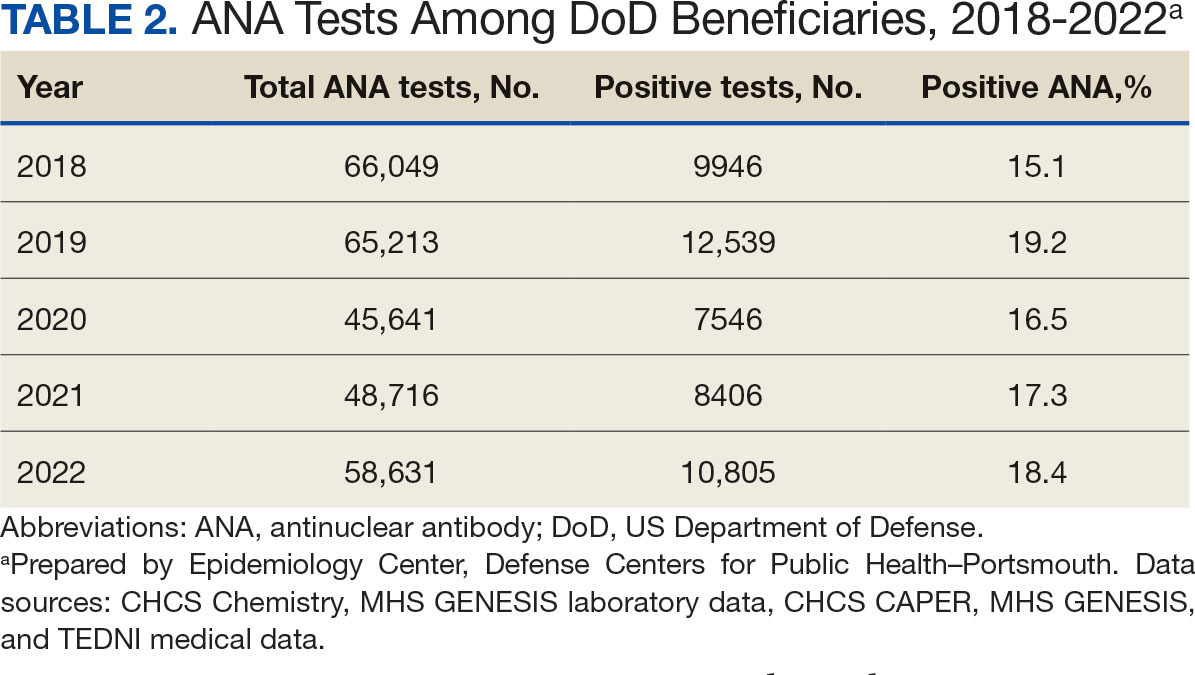
A mean 56,850 ANA tests were conducted annually in centralized laboratories using standardized protocols (Table 2). The mean ANA positivity rate was 17.3%, which remained relatively stable from 2018 through 2022.
Discussion
This study examined the annual incidence of newly diagnosed SLE cases among all TRICARE beneficiaries from January 1, 2018, through December 31, 2022, covering both before and during the peak years of the COVID-19 pandemic. This analysis revealed a steady decline in SLE cases during this period. The reliability of these findings is reinforced by the comprehensiveness of the MHS, one of the largest US health care delivery systems, which maintains near-complete medical data capture for about 9.5 million DoD TRICARE beneficiaries across domestic and international settings.
SLE is a rare autoimmune disorder that presents a diagnostic challenge due to its wide range of nonspecific symptoms, many of which resemble other conditions. To reduce the likelihood of false-positive results and ensure diagnostic accuracy, this study adopted a stringent case definition. Incident cases were identified by the presence of ANA testing in conjunction with lupus-specific ICD-10-CM codes and required ≥ 4 lupus related diagnostic entries. This criterion was necessary due to the absence of ANA test results in data from private sector care settings. Our case definition aligns with established literature. For example, a Vanderbilt University chart review study demonstrated that combining ANA positivity with ≥ 4 lupus related ICD-10-CM codes achieves a positive predictive value of 100%, albeit with a sensitivity of 45%.11 Other studies similarly affirm the diagnostic validity of using recurrent ICD-10-CM codes to improve specificity in identifying lupus cases.12,13
The primary objective of this study was to examine the temporal trend in newly diagnosed lupus cases, rather than derive precise incidence rates. Although the TRICARE system includes about 9.5 million beneficiaries, this number represents a dynamic population with continual inflow and outflow. Accurate incidence rate calculation would require access to detailed denominator data, which were not readily available. In comparison with our findings, a study limited to active-duty service members reported fewer lupus cases. This discrepancy likely reflects differences in case definitions—specifically, the absence of laboratory data, the restricted range of diagnostic codes, and the requirement that diagnoses be rendered by specialists.14 Despite these differences, demographic patterns were consistent, with higher incidence observed in females and individuals aged ≥ 20 years.
A Centers for Disease Control and Prevention (CDC) study of lupus incidence in the general population also reported lower case counts.1 However, the CDC estimates were based on 5 state-level registries, which rely on clinician-reported cases and therefore may underestimate true disease burden. Moreover, the DoD beneficiary population differs markedly from the general population: it includes a large cohort of retirees, ensuring an older demographic; all members have comprehensive health care access; and active-duty personnel are subject to pre-enlistment medical screening. Taken together, these factors suggest this study may offer a more complete and systematically captured profile of lupus incidence.
We observed a marked decline of newly diagnosed SLE cases during the study period, which coincided with the widespread circulation of COVID-19. This decrease is unlikely to be attributable to reduced access to care during the pandemic. The MHS operates under a single-payer model, and the total number of patient encounters remained relatively stable throughout the pandemic.
To our knowledge, this is the only study to monitor lupus incidence in a large US population over the 5-year period encompassing before and during the COVID-19 pandemic. To date, only 4 large-scale surveillance studies have addressed similar questions. 14-17 Our findings are consistent with the most recent of these reports: an analysis limited to active-duty members of the US Armed Forces identified 1127 patients with newly diagnosed lupus between 2000 and 2022 and reported stable incidence trends throughout the pandemic.14 The other 3 studies adopted a different approach, comparing the emergence of autoimmune diseases, including lupus, between individuals with confirmed SARS-CoV-2 infection and those without. Each of these trials concluded that COVID-19 increases the risk of various autoimmune conditions, although the findings specific to lupus were inconsistent.15-17
Chang et al reported a significant increase in new lupus diagnoses (n = 2,926,016), with an adjusted hazard ratio (aHR) of 2.99 (95% CI, 2.68-3.34), spanning all ages and both sexes. The highest incidence was observed in individuals of Asian descent.15 Using German routine health care data from 2020, Tesch et al identified a heightened risk of autoimmune diseases, including lupus, among patients with a history of SARS-CoV-2 infection (n = 641,407; 9.4% children, 57.3% female, 6.4% hospitalized), compared with matched infection-naïve controls (n = 1,560,357).16 Both studies excluded vaccinated individuals.
These 2 studies diverged in their assessment of the relationship between COVID-19 severity and subsequent autoimmune risk. Chang et al found a higher incidence among nonhospitalized ambulatory patients, while Tesch et al reported that increased risk was associated with patients requiring intensive care unit admission.15,16
In contrast, based on a cohort of 4,197,188 individuals, Peng et al found no significant difference in lupus incidence among patients with SARS-CoV-2 infection (aHR, 1.05; 95% CI, 0.79-1.39).17 Notably, within the infected group, the incidence of SLE was significantly lower among vaccinated individuals compared with the unvaccinated group (aHR, 0.29; 95% CI, 0.18-0.47). Similar protective associations were observed for other antibody-mediated autoimmune disorders, including pemphigoid, Graves’ disease, and antiphospholipid antibody syndrome.
Limitations
Due to fundamental differences in study design, we were unable to directly reconcile our findings with those reported in the literature. This study lacked access to reliable documentation of COVID-19 infection status, primarily due to the widespread use of home testing among TRICARE beneficiaries. Additionally, the dataset did not include inpatient records and therefore did not permit evaluation of disease severity. Despite these constraints, it is plausible that the overall burden of COVID-19 infection within the study population was lower than that observed in the general US population.
As of December 2022, the DoD had reported about 750,000 confirmed COVID-19 cases among military personnel, civilian employees, dependents, and DoD contractors.18 Given that TRICARE beneficiaries represent about 2.8% of the total US population—and that > 90 million US individuals were infected between 2020 and 2022—the implied infection rate in our cohort appears to be about one-third of what might be expected.19 This discrepancy may be due to higher adherence to mitigation measures, such as social distancing and mask usage, among DoD-affiliated populations. COVID-19 vaccination was mandated for all active-duty service members, who constitute 5.4% of the study population. The remaining TRICARE beneficiaries also had access to guaranteed health care and vaccination coverage, likely contributing to high overall vaccination rates.
Because > 80% of the study population was composed of individuals from diverse civilian backgrounds, we expect the distribution of infection severity within the DoD beneficiary population to approximate that of the general US population.
Future Directions
The findings of this study offer circumstantial yet real-time evidence of the complexity underlying immune dysregulation at the intersection of host susceptibility and environmental exposures. The stability in ANA positivity rates during the study period mitigates concerns regarding undiagnosed subclinical lupus and may suggest that, overall, immune homeostasis was preserved among DoD beneficiaries.
It is noteworthy that during the COVID-19 pandemic, the incidence of several common infections—such as influenza and EBV—declined markedly, likely as a result of widespread social distancing and other public health interventions.20 Mitigation strategies implemented within the military may have conferred protection not only against COVID-19 but also against other community-acquired pathogens.
In light of these observations, we hypothesize that for COVID-19 to act as a trigger for SLE, a prolonged or repeated disruption of immune equilibrium may be required—potentially mediated by recurrent infections or sustained inflammatory states. The association between viral infections and autoimmunity is well established. Immune dysregulation leading to autoantibody production has been observed not only in the context of SARS-CoV-2 but also following infections with EBV, cytomegalovirus, enteroviruses, hepatitis B and C viruses, HIV, and parvovirus B19.21
This dysregulation is often transient, accompanied by compensatory immune regulatory responses. However, in individuals subjected to successive or overlapping infections, these regulatory mechanisms may become compromised or overwhelmed, due to emergent patterns of immune interference of varying severity. In such cases, a transient immune perturbation may progress into a bona fide autoimmune disease, contingent upon individual risk factors such as genetic predisposition, preexisting immune memory, and regenerative capacity.21
Therefore, we believe the significance of this study is 2-fold. First, lupus is known to develop gradually and may require 3 to 5 years to clinically manifest after the initial break in immunological tolerance.3 Continued public health surveillance represents a more pragmatic strategy than retrospective cohort construction, especially as histories of COVID-19 infection become increasingly complete and definitive. Our findings provide a valuable baseline reference point for future longitudinal studies.
The interpretation of surveillance outcomes—whether indicating an upward trend, a stable baseline, or a downward trend—offers distinct analytical value. Within this study population, we observed neither an upward trajectory that might suggest a direct causal link, nor a flat trend that would imply absence of association between COVID-19 and lupus pathogenesis. Instead, the observation of a downward trend invites consideration of nonlinear or protective influences. From this perspective, we recommend that future investigations adopt a holistic framework when assessing environmental contributions to immune dysregulation—particularly when evaluating the long-term immunopathological consequences of the COVID-19 pandemic on lupus and related autoimmune conditions.
Conclusions
This study identified a declining trend in incident lupus cases during the COVID-19 pandemic among the DoD beneficiary population. Further investigation is warranted to elucidate the underlying factors contributing to this decline. Conducting longitudinal epidemiologic studies and applying multivariable regression analyses will be essential to determine whether incidence rates revert to prepandemic baselines and how these trends may be influenced by evolving environmental factors within the general population.
Systemic lupus erythematosus (SLE), or lupus, is a rare autoimmune disease estimated to occur in about 5.1 cases per 100,000 person-years in the United States in 2018.1 The disease predominantly affects females, with an incidence of 8.7 cases per 100,000 person-years vs 1.2 cases per 100,000 person-years in males, and is most common in patients aged 15 to 44 years.1,2
Lupus presents with a constellation of clinical signs and symptoms that evolve, along with hallmark laboratory findings indicative of immune dysregulation and polyclonal B-cell activation. Consequently, a wide array of autoantibodies may be produced, although the combination of epitope specificity can vary from patient to patient.3 Nevertheless, > 98% of individuals diagnosed with lupus produce antinuclear antibodies (ANA), making ANA positivity a near-universal serologic feature at the time of diagnosis.
The pathogenesis of lupus is complex. Research from the past 5 decades supports the role of certain viral infections—such as Epstein-Barr virus (EBV) and cytomegalovirus—as potential triggers.4 These viruses are thought to initiate disease through mechanisms including activation of interferon pathways, exposure of cryptic intracellular antigens, molecular mimicry, and epitope spreading. Subsequent clonal expansion and autoantibody production occur to varying degrees, influenced by viral load and host susceptibility factors.
During the COVID-19 pandemic, it became evident that SARS-CoV-2 exerts profound effects on immune regulation, influencing infection outcomes through mechanisms such as hyperactivation of innate immunity, especially in the lungs, leading to acute respiratory distress syndrome. Additionally, SARS-CoV-2 has been associated with polyclonal B-cell activation and the generation of autoantibodies. This association gained attention after Bastard et al identified anti–type I interferon antibodies in patients with severe COVID-19, predominantly among males with a genetic predisposition. These autoantibodies were shown to impair antiviral defenses and contribute to life-threatening pneumonia.5
Subsequent studies demonstrated the production of a wide spectrum of functional autoantibodies, including ANA, in patients with COVID-19.6,7 These findings were attributed to the acute expansion of autoreactive clones among naïve-derived immunoglobulin G1 antibody-secreting cells during the early stages of infection, with the degree of expansion correlating with disease severity.8,9 Although longitudinal data up to 15 months postinfection suggest this serologic abnormality resolves in more than two-thirds of patients, the number of individuals infected globally has raised serious public health concerns regarding the potential long-term sequelae, including the onset of lupus or other autoimmune diseases in COVID-19 survivors.6-9 A limited number of case reports describing the onset of lupus following SARS-CoV-2 infection support this hypothesis.10
This surveillance analysis investigates lupus incidence among patients within the Military Health System (MHS), encompassing all TRICARE beneficiaries, from January 2018 to December 2022. The objective of this analysis was to examine lupus incidence trends throughout the COVID-19 pandemic, stratified by sex, age, and active-duty status.
Methods
The MHS provides health care services to about 9.5 million US Department of Defense (DoD) beneficiaries. Outpatient health records and laboratory results for individuals receiving care at military treatment facilities (MTFs) between January 1, 2018, and December 31, 2022, were obtained from the Comprehensive Ambulatory/ Professional Encounter Record and MHS GENESIS. For beneficiaries receiving care in the private sector, data were sourced from the TRICARE Encounter Data—Non-Institutional database.
Laboratory test results, including ANA testing, were available only for individuals receiving care at MTFs. These laboratory data were extracted from the Composite Health Care System Chemistry database and MHS GENESIS laboratory systems for the same time frame. Inpatient data were not included in this analysis. Data from 2017 were used solely as a look-back (or washout) period to identify and exclude prevalent lupus cases diagnosed before 2018 and were not included in the final results.
Lupus cases were identified by the presence of a positive ANA test and appropriate International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) codes. A positive ANA result was defined as either a qualitative result marked positive or a titer ≥ 1:80. The ICD-10-CM codes considered indicative of lupus included variations of M32, L93, or H01.12.
M32, L93, or H01.12. For cases with a positive ANA test, a lupus diagnosis required the presence of ≥ 2 lupus related ICD-10-CM codes. In the absence of ANA test results, a stricter criterion was applied: ≥ 4 lupus ICD-10-CM diagnosis codes recorded on separate days were required for inclusion.
Beneficiaries were excluded if they had a negative ANA result, only 1 lupus ICD- 10-CM diagnosis code, 1 positive ANA with only 1 corresponding ICD-10-CM code, or if their diagnosis occurred outside the defined study period. Patients and members of the public were not involved in the design, conduct, reporting, or dissemination of this study.
Results
Between January 1, 2017, and December 31, 2022, 99,946 TRICARE beneficiaries had some indication of lupus testing or diagnosis in their health records (Figure 1). Of these beneficiaries, 5335 had a positive ANA result and ≥ 2 ICD-10-CM lupus diagnosis codes. An additional 28,275 beneficiaries had ≥ 4 ICD-10-CM lupus diagnosis codes but no ANA test results. From these groups, the final sample included 10,760 beneficiaries who met the incident case definitions for SLE during the study period (2018 through 2022).
Most cases (85.1%, n = 9157) were diagnosed through TRICARE claims, while 1205 (11.2%) were diagnosed within the MHS. Another 398 (3.7%) had documentation of care both within and outside the MHS. Incident SLE cases declined by an average of 16% annually during the study period (Figure 2). This trend amounted to an overall reduction of 48.2%, from 2866 cases in 2018 to 1399 cases in 2022. This decline occurred despite total medical encounters among DoD beneficiaries remaining relatively stable during the pandemic years, with only a 3.5% change between 2018 and 2022.
The disease was more prevalent among female beneficiaries, with a female to- male ratio of 7:1 (Table 1). Among women, the number of new cases declined from 2519 in 2018 to 1223 in 2022, while the number of cases among men remained consistently < 350 annually. Similar trends were observed across other strata. Incident SLE cases were more common among nonactive-duty beneficiaries than active-duty service members, with a ratio of 18:1. New cases among active-duty members remained < 155 per year. Age-stratified data revealed that SLE was diagnosed predominantly in individuals aged ≥ 18 years, with a ratio of 37:1 compared with individuals aged < 18 years. Among children, the number of new cases remained < 75 per year throughout the study period.
A mean 56,850 ANA tests were conducted annually in centralized laboratories using standardized protocols (Table 2). The mean ANA positivity rate was 17.3%, which remained relatively stable from 2018 through 2022.
Discussion
This study examined the annual incidence of newly diagnosed SLE cases among all TRICARE beneficiaries from January 1, 2018, through December 31, 2022, covering both before and during the peak years of the COVID-19 pandemic. This analysis revealed a steady decline in SLE cases during this period. The reliability of these findings is reinforced by the comprehensiveness of the MHS, one of the largest US health care delivery systems, which maintains near-complete medical data capture for about 9.5 million DoD TRICARE beneficiaries across domestic and international settings.
SLE is a rare autoimmune disorder that presents a diagnostic challenge due to its wide range of nonspecific symptoms, many of which resemble other conditions. To reduce the likelihood of false-positive results and ensure diagnostic accuracy, this study adopted a stringent case definition. Incident cases were identified by the presence of ANA testing in conjunction with lupus-specific ICD-10-CM codes and required ≥ 4 lupus related diagnostic entries. This criterion was necessary due to the absence of ANA test results in data from private sector care settings. Our case definition aligns with established literature. For example, a Vanderbilt University chart review study demonstrated that combining ANA positivity with ≥ 4 lupus related ICD-10-CM codes achieves a positive predictive value of 100%, albeit with a sensitivity of 45%.11 Other studies similarly affirm the diagnostic validity of using recurrent ICD-10-CM codes to improve specificity in identifying lupus cases.12,13
The primary objective of this study was to examine the temporal trend in newly diagnosed lupus cases, rather than derive precise incidence rates. Although the TRICARE system includes about 9.5 million beneficiaries, this number represents a dynamic population with continual inflow and outflow. Accurate incidence rate calculation would require access to detailed denominator data, which were not readily available. In comparison with our findings, a study limited to active-duty service members reported fewer lupus cases. This discrepancy likely reflects differences in case definitions—specifically, the absence of laboratory data, the restricted range of diagnostic codes, and the requirement that diagnoses be rendered by specialists.14 Despite these differences, demographic patterns were consistent, with higher incidence observed in females and individuals aged ≥ 20 years.
A Centers for Disease Control and Prevention (CDC) study of lupus incidence in the general population also reported lower case counts.1 However, the CDC estimates were based on 5 state-level registries, which rely on clinician-reported cases and therefore may underestimate true disease burden. Moreover, the DoD beneficiary population differs markedly from the general population: it includes a large cohort of retirees, ensuring an older demographic; all members have comprehensive health care access; and active-duty personnel are subject to pre-enlistment medical screening. Taken together, these factors suggest this study may offer a more complete and systematically captured profile of lupus incidence.
We observed a marked decline of newly diagnosed SLE cases during the study period, which coincided with the widespread circulation of COVID-19. This decrease is unlikely to be attributable to reduced access to care during the pandemic. The MHS operates under a single-payer model, and the total number of patient encounters remained relatively stable throughout the pandemic.
To our knowledge, this is the only study to monitor lupus incidence in a large US population over the 5-year period encompassing before and during the COVID-19 pandemic. To date, only 4 large-scale surveillance studies have addressed similar questions. 14-17 Our findings are consistent with the most recent of these reports: an analysis limited to active-duty members of the US Armed Forces identified 1127 patients with newly diagnosed lupus between 2000 and 2022 and reported stable incidence trends throughout the pandemic.14 The other 3 studies adopted a different approach, comparing the emergence of autoimmune diseases, including lupus, between individuals with confirmed SARS-CoV-2 infection and those without. Each of these trials concluded that COVID-19 increases the risk of various autoimmune conditions, although the findings specific to lupus were inconsistent.15-17
Chang et al reported a significant increase in new lupus diagnoses (n = 2,926,016), with an adjusted hazard ratio (aHR) of 2.99 (95% CI, 2.68-3.34), spanning all ages and both sexes. The highest incidence was observed in individuals of Asian descent.15 Using German routine health care data from 2020, Tesch et al identified a heightened risk of autoimmune diseases, including lupus, among patients with a history of SARS-CoV-2 infection (n = 641,407; 9.4% children, 57.3% female, 6.4% hospitalized), compared with matched infection-naïve controls (n = 1,560,357).16 Both studies excluded vaccinated individuals.
These 2 studies diverged in their assessment of the relationship between COVID-19 severity and subsequent autoimmune risk. Chang et al found a higher incidence among nonhospitalized ambulatory patients, while Tesch et al reported that increased risk was associated with patients requiring intensive care unit admission.15,16
In contrast, based on a cohort of 4,197,188 individuals, Peng et al found no significant difference in lupus incidence among patients with SARS-CoV-2 infection (aHR, 1.05; 95% CI, 0.79-1.39).17 Notably, within the infected group, the incidence of SLE was significantly lower among vaccinated individuals compared with the unvaccinated group (aHR, 0.29; 95% CI, 0.18-0.47). Similar protective associations were observed for other antibody-mediated autoimmune disorders, including pemphigoid, Graves’ disease, and antiphospholipid antibody syndrome.
Limitations
Due to fundamental differences in study design, we were unable to directly reconcile our findings with those reported in the literature. This study lacked access to reliable documentation of COVID-19 infection status, primarily due to the widespread use of home testing among TRICARE beneficiaries. Additionally, the dataset did not include inpatient records and therefore did not permit evaluation of disease severity. Despite these constraints, it is plausible that the overall burden of COVID-19 infection within the study population was lower than that observed in the general US population.
As of December 2022, the DoD had reported about 750,000 confirmed COVID-19 cases among military personnel, civilian employees, dependents, and DoD contractors.18 Given that TRICARE beneficiaries represent about 2.8% of the total US population—and that > 90 million US individuals were infected between 2020 and 2022—the implied infection rate in our cohort appears to be about one-third of what might be expected.19 This discrepancy may be due to higher adherence to mitigation measures, such as social distancing and mask usage, among DoD-affiliated populations. COVID-19 vaccination was mandated for all active-duty service members, who constitute 5.4% of the study population. The remaining TRICARE beneficiaries also had access to guaranteed health care and vaccination coverage, likely contributing to high overall vaccination rates.
Because > 80% of the study population was composed of individuals from diverse civilian backgrounds, we expect the distribution of infection severity within the DoD beneficiary population to approximate that of the general US population.
Future Directions
The findings of this study offer circumstantial yet real-time evidence of the complexity underlying immune dysregulation at the intersection of host susceptibility and environmental exposures. The stability in ANA positivity rates during the study period mitigates concerns regarding undiagnosed subclinical lupus and may suggest that, overall, immune homeostasis was preserved among DoD beneficiaries.
It is noteworthy that during the COVID-19 pandemic, the incidence of several common infections—such as influenza and EBV—declined markedly, likely as a result of widespread social distancing and other public health interventions.20 Mitigation strategies implemented within the military may have conferred protection not only against COVID-19 but also against other community-acquired pathogens.
In light of these observations, we hypothesize that for COVID-19 to act as a trigger for SLE, a prolonged or repeated disruption of immune equilibrium may be required—potentially mediated by recurrent infections or sustained inflammatory states. The association between viral infections and autoimmunity is well established. Immune dysregulation leading to autoantibody production has been observed not only in the context of SARS-CoV-2 but also following infections with EBV, cytomegalovirus, enteroviruses, hepatitis B and C viruses, HIV, and parvovirus B19.21
This dysregulation is often transient, accompanied by compensatory immune regulatory responses. However, in individuals subjected to successive or overlapping infections, these regulatory mechanisms may become compromised or overwhelmed, due to emergent patterns of immune interference of varying severity. In such cases, a transient immune perturbation may progress into a bona fide autoimmune disease, contingent upon individual risk factors such as genetic predisposition, preexisting immune memory, and regenerative capacity.21
Therefore, we believe the significance of this study is 2-fold. First, lupus is known to develop gradually and may require 3 to 5 years to clinically manifest after the initial break in immunological tolerance.3 Continued public health surveillance represents a more pragmatic strategy than retrospective cohort construction, especially as histories of COVID-19 infection become increasingly complete and definitive. Our findings provide a valuable baseline reference point for future longitudinal studies.
The interpretation of surveillance outcomes—whether indicating an upward trend, a stable baseline, or a downward trend—offers distinct analytical value. Within this study population, we observed neither an upward trajectory that might suggest a direct causal link, nor a flat trend that would imply absence of association between COVID-19 and lupus pathogenesis. Instead, the observation of a downward trend invites consideration of nonlinear or protective influences. From this perspective, we recommend that future investigations adopt a holistic framework when assessing environmental contributions to immune dysregulation—particularly when evaluating the long-term immunopathological consequences of the COVID-19 pandemic on lupus and related autoimmune conditions.
Conclusions
This study identified a declining trend in incident lupus cases during the COVID-19 pandemic among the DoD beneficiary population. Further investigation is warranted to elucidate the underlying factors contributing to this decline. Conducting longitudinal epidemiologic studies and applying multivariable regression analyses will be essential to determine whether incidence rates revert to prepandemic baselines and how these trends may be influenced by evolving environmental factors within the general population.
Systemic lupus erythematosus (SLE), or lupus, is a rare autoimmune disease estimated to occur in about 5.1 cases per 100,000 person-years in the United States in 2018.1 The disease predominantly affects females, with an incidence of 8.7 cases per 100,000 person-years vs 1.2 cases per 100,000 person-years in males, and is most common in patients aged 15 to 44 years.1,2
Lupus presents with a constellation of clinical signs and symptoms that evolve, along with hallmark laboratory findings indicative of immune dysregulation and polyclonal B-cell activation. Consequently, a wide array of autoantibodies may be produced, although the combination of epitope specificity can vary from patient to patient.3 Nevertheless, > 98% of individuals diagnosed with lupus produce antinuclear antibodies (ANA), making ANA positivity a near-universal serologic feature at the time of diagnosis.
The pathogenesis of lupus is complex. Research from the past 5 decades supports the role of certain viral infections—such as Epstein-Barr virus (EBV) and cytomegalovirus—as potential triggers.4 These viruses are thought to initiate disease through mechanisms including activation of interferon pathways, exposure of cryptic intracellular antigens, molecular mimicry, and epitope spreading. Subsequent clonal expansion and autoantibody production occur to varying degrees, influenced by viral load and host susceptibility factors.
During the COVID-19 pandemic, it became evident that SARS-CoV-2 exerts profound effects on immune regulation, influencing infection outcomes through mechanisms such as hyperactivation of innate immunity, especially in the lungs, leading to acute respiratory distress syndrome. Additionally, SARS-CoV-2 has been associated with polyclonal B-cell activation and the generation of autoantibodies. This association gained attention after Bastard et al identified anti–type I interferon antibodies in patients with severe COVID-19, predominantly among males with a genetic predisposition. These autoantibodies were shown to impair antiviral defenses and contribute to life-threatening pneumonia.5
Subsequent studies demonstrated the production of a wide spectrum of functional autoantibodies, including ANA, in patients with COVID-19.6,7 These findings were attributed to the acute expansion of autoreactive clones among naïve-derived immunoglobulin G1 antibody-secreting cells during the early stages of infection, with the degree of expansion correlating with disease severity.8,9 Although longitudinal data up to 15 months postinfection suggest this serologic abnormality resolves in more than two-thirds of patients, the number of individuals infected globally has raised serious public health concerns regarding the potential long-term sequelae, including the onset of lupus or other autoimmune diseases in COVID-19 survivors.6-9 A limited number of case reports describing the onset of lupus following SARS-CoV-2 infection support this hypothesis.10
This surveillance analysis investigates lupus incidence among patients within the Military Health System (MHS), encompassing all TRICARE beneficiaries, from January 2018 to December 2022. The objective of this analysis was to examine lupus incidence trends throughout the COVID-19 pandemic, stratified by sex, age, and active-duty status.
Methods
The MHS provides health care services to about 9.5 million US Department of Defense (DoD) beneficiaries. Outpatient health records and laboratory results for individuals receiving care at military treatment facilities (MTFs) between January 1, 2018, and December 31, 2022, were obtained from the Comprehensive Ambulatory/ Professional Encounter Record and MHS GENESIS. For beneficiaries receiving care in the private sector, data were sourced from the TRICARE Encounter Data—Non-Institutional database.
Laboratory test results, including ANA testing, were available only for individuals receiving care at MTFs. These laboratory data were extracted from the Composite Health Care System Chemistry database and MHS GENESIS laboratory systems for the same time frame. Inpatient data were not included in this analysis. Data from 2017 were used solely as a look-back (or washout) period to identify and exclude prevalent lupus cases diagnosed before 2018 and were not included in the final results.
Lupus cases were identified by the presence of a positive ANA test and appropriate International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) codes. A positive ANA result was defined as either a qualitative result marked positive or a titer ≥ 1:80. The ICD-10-CM codes considered indicative of lupus included variations of M32, L93, or H01.12.
M32, L93, or H01.12. For cases with a positive ANA test, a lupus diagnosis required the presence of ≥ 2 lupus related ICD-10-CM codes. In the absence of ANA test results, a stricter criterion was applied: ≥ 4 lupus ICD-10-CM diagnosis codes recorded on separate days were required for inclusion.
Beneficiaries were excluded if they had a negative ANA result, only 1 lupus ICD- 10-CM diagnosis code, 1 positive ANA with only 1 corresponding ICD-10-CM code, or if their diagnosis occurred outside the defined study period. Patients and members of the public were not involved in the design, conduct, reporting, or dissemination of this study.
Results
Between January 1, 2017, and December 31, 2022, 99,946 TRICARE beneficiaries had some indication of lupus testing or diagnosis in their health records (Figure 1). Of these beneficiaries, 5335 had a positive ANA result and ≥ 2 ICD-10-CM lupus diagnosis codes. An additional 28,275 beneficiaries had ≥ 4 ICD-10-CM lupus diagnosis codes but no ANA test results. From these groups, the final sample included 10,760 beneficiaries who met the incident case definitions for SLE during the study period (2018 through 2022).
Most cases (85.1%, n = 9157) were diagnosed through TRICARE claims, while 1205 (11.2%) were diagnosed within the MHS. Another 398 (3.7%) had documentation of care both within and outside the MHS. Incident SLE cases declined by an average of 16% annually during the study period (Figure 2). This trend amounted to an overall reduction of 48.2%, from 2866 cases in 2018 to 1399 cases in 2022. This decline occurred despite total medical encounters among DoD beneficiaries remaining relatively stable during the pandemic years, with only a 3.5% change between 2018 and 2022.
The disease was more prevalent among female beneficiaries, with a female to- male ratio of 7:1 (Table 1). Among women, the number of new cases declined from 2519 in 2018 to 1223 in 2022, while the number of cases among men remained consistently < 350 annually. Similar trends were observed across other strata. Incident SLE cases were more common among nonactive-duty beneficiaries than active-duty service members, with a ratio of 18:1. New cases among active-duty members remained < 155 per year. Age-stratified data revealed that SLE was diagnosed predominantly in individuals aged ≥ 18 years, with a ratio of 37:1 compared with individuals aged < 18 years. Among children, the number of new cases remained < 75 per year throughout the study period.
A mean 56,850 ANA tests were conducted annually in centralized laboratories using standardized protocols (Table 2). The mean ANA positivity rate was 17.3%, which remained relatively stable from 2018 through 2022.
Discussion
This study examined the annual incidence of newly diagnosed SLE cases among all TRICARE beneficiaries from January 1, 2018, through December 31, 2022, covering both before and during the peak years of the COVID-19 pandemic. This analysis revealed a steady decline in SLE cases during this period. The reliability of these findings is reinforced by the comprehensiveness of the MHS, one of the largest US health care delivery systems, which maintains near-complete medical data capture for about 9.5 million DoD TRICARE beneficiaries across domestic and international settings.
SLE is a rare autoimmune disorder that presents a diagnostic challenge due to its wide range of nonspecific symptoms, many of which resemble other conditions. To reduce the likelihood of false-positive results and ensure diagnostic accuracy, this study adopted a stringent case definition. Incident cases were identified by the presence of ANA testing in conjunction with lupus-specific ICD-10-CM codes and required ≥ 4 lupus related diagnostic entries. This criterion was necessary due to the absence of ANA test results in data from private sector care settings. Our case definition aligns with established literature. For example, a Vanderbilt University chart review study demonstrated that combining ANA positivity with ≥ 4 lupus related ICD-10-CM codes achieves a positive predictive value of 100%, albeit with a sensitivity of 45%.11 Other studies similarly affirm the diagnostic validity of using recurrent ICD-10-CM codes to improve specificity in identifying lupus cases.12,13
The primary objective of this study was to examine the temporal trend in newly diagnosed lupus cases, rather than derive precise incidence rates. Although the TRICARE system includes about 9.5 million beneficiaries, this number represents a dynamic population with continual inflow and outflow. Accurate incidence rate calculation would require access to detailed denominator data, which were not readily available. In comparison with our findings, a study limited to active-duty service members reported fewer lupus cases. This discrepancy likely reflects differences in case definitions—specifically, the absence of laboratory data, the restricted range of diagnostic codes, and the requirement that diagnoses be rendered by specialists.14 Despite these differences, demographic patterns were consistent, with higher incidence observed in females and individuals aged ≥ 20 years.
A Centers for Disease Control and Prevention (CDC) study of lupus incidence in the general population also reported lower case counts.1 However, the CDC estimates were based on 5 state-level registries, which rely on clinician-reported cases and therefore may underestimate true disease burden. Moreover, the DoD beneficiary population differs markedly from the general population: it includes a large cohort of retirees, ensuring an older demographic; all members have comprehensive health care access; and active-duty personnel are subject to pre-enlistment medical screening. Taken together, these factors suggest this study may offer a more complete and systematically captured profile of lupus incidence.
We observed a marked decline of newly diagnosed SLE cases during the study period, which coincided with the widespread circulation of COVID-19. This decrease is unlikely to be attributable to reduced access to care during the pandemic. The MHS operates under a single-payer model, and the total number of patient encounters remained relatively stable throughout the pandemic.
To our knowledge, this is the only study to monitor lupus incidence in a large US population over the 5-year period encompassing before and during the COVID-19 pandemic. To date, only 4 large-scale surveillance studies have addressed similar questions. 14-17 Our findings are consistent with the most recent of these reports: an analysis limited to active-duty members of the US Armed Forces identified 1127 patients with newly diagnosed lupus between 2000 and 2022 and reported stable incidence trends throughout the pandemic.14 The other 3 studies adopted a different approach, comparing the emergence of autoimmune diseases, including lupus, between individuals with confirmed SARS-CoV-2 infection and those without. Each of these trials concluded that COVID-19 increases the risk of various autoimmune conditions, although the findings specific to lupus were inconsistent.15-17
Chang et al reported a significant increase in new lupus diagnoses (n = 2,926,016), with an adjusted hazard ratio (aHR) of 2.99 (95% CI, 2.68-3.34), spanning all ages and both sexes. The highest incidence was observed in individuals of Asian descent.15 Using German routine health care data from 2020, Tesch et al identified a heightened risk of autoimmune diseases, including lupus, among patients with a history of SARS-CoV-2 infection (n = 641,407; 9.4% children, 57.3% female, 6.4% hospitalized), compared with matched infection-naïve controls (n = 1,560,357).16 Both studies excluded vaccinated individuals.
These 2 studies diverged in their assessment of the relationship between COVID-19 severity and subsequent autoimmune risk. Chang et al found a higher incidence among nonhospitalized ambulatory patients, while Tesch et al reported that increased risk was associated with patients requiring intensive care unit admission.15,16
In contrast, based on a cohort of 4,197,188 individuals, Peng et al found no significant difference in lupus incidence among patients with SARS-CoV-2 infection (aHR, 1.05; 95% CI, 0.79-1.39).17 Notably, within the infected group, the incidence of SLE was significantly lower among vaccinated individuals compared with the unvaccinated group (aHR, 0.29; 95% CI, 0.18-0.47). Similar protective associations were observed for other antibody-mediated autoimmune disorders, including pemphigoid, Graves’ disease, and antiphospholipid antibody syndrome.
Limitations
Due to fundamental differences in study design, we were unable to directly reconcile our findings with those reported in the literature. This study lacked access to reliable documentation of COVID-19 infection status, primarily due to the widespread use of home testing among TRICARE beneficiaries. Additionally, the dataset did not include inpatient records and therefore did not permit evaluation of disease severity. Despite these constraints, it is plausible that the overall burden of COVID-19 infection within the study population was lower than that observed in the general US population.
As of December 2022, the DoD had reported about 750,000 confirmed COVID-19 cases among military personnel, civilian employees, dependents, and DoD contractors.18 Given that TRICARE beneficiaries represent about 2.8% of the total US population—and that > 90 million US individuals were infected between 2020 and 2022—the implied infection rate in our cohort appears to be about one-third of what might be expected.19 This discrepancy may be due to higher adherence to mitigation measures, such as social distancing and mask usage, among DoD-affiliated populations. COVID-19 vaccination was mandated for all active-duty service members, who constitute 5.4% of the study population. The remaining TRICARE beneficiaries also had access to guaranteed health care and vaccination coverage, likely contributing to high overall vaccination rates.
Because > 80% of the study population was composed of individuals from diverse civilian backgrounds, we expect the distribution of infection severity within the DoD beneficiary population to approximate that of the general US population.
Future Directions
The findings of this study offer circumstantial yet real-time evidence of the complexity underlying immune dysregulation at the intersection of host susceptibility and environmental exposures. The stability in ANA positivity rates during the study period mitigates concerns regarding undiagnosed subclinical lupus and may suggest that, overall, immune homeostasis was preserved among DoD beneficiaries.
It is noteworthy that during the COVID-19 pandemic, the incidence of several common infections—such as influenza and EBV—declined markedly, likely as a result of widespread social distancing and other public health interventions.20 Mitigation strategies implemented within the military may have conferred protection not only against COVID-19 but also against other community-acquired pathogens.
In light of these observations, we hypothesize that for COVID-19 to act as a trigger for SLE, a prolonged or repeated disruption of immune equilibrium may be required—potentially mediated by recurrent infections or sustained inflammatory states. The association between viral infections and autoimmunity is well established. Immune dysregulation leading to autoantibody production has been observed not only in the context of SARS-CoV-2 but also following infections with EBV, cytomegalovirus, enteroviruses, hepatitis B and C viruses, HIV, and parvovirus B19.21
This dysregulation is often transient, accompanied by compensatory immune regulatory responses. However, in individuals subjected to successive or overlapping infections, these regulatory mechanisms may become compromised or overwhelmed, due to emergent patterns of immune interference of varying severity. In such cases, a transient immune perturbation may progress into a bona fide autoimmune disease, contingent upon individual risk factors such as genetic predisposition, preexisting immune memory, and regenerative capacity.21
Therefore, we believe the significance of this study is 2-fold. First, lupus is known to develop gradually and may require 3 to 5 years to clinically manifest after the initial break in immunological tolerance.3 Continued public health surveillance represents a more pragmatic strategy than retrospective cohort construction, especially as histories of COVID-19 infection become increasingly complete and definitive. Our findings provide a valuable baseline reference point for future longitudinal studies.
The interpretation of surveillance outcomes—whether indicating an upward trend, a stable baseline, or a downward trend—offers distinct analytical value. Within this study population, we observed neither an upward trajectory that might suggest a direct causal link, nor a flat trend that would imply absence of association between COVID-19 and lupus pathogenesis. Instead, the observation of a downward trend invites consideration of nonlinear or protective influences. From this perspective, we recommend that future investigations adopt a holistic framework when assessing environmental contributions to immune dysregulation—particularly when evaluating the long-term immunopathological consequences of the COVID-19 pandemic on lupus and related autoimmune conditions.
Conclusions
This study identified a declining trend in incident lupus cases during the COVID-19 pandemic among the DoD beneficiary population. Further investigation is warranted to elucidate the underlying factors contributing to this decline. Conducting longitudinal epidemiologic studies and applying multivariable regression analyses will be essential to determine whether incidence rates revert to prepandemic baselines and how these trends may be influenced by evolving environmental factors within the general population.
A Systemic Lupus Erythematosus Incidence Surveillance Report Among DoD Beneficiaries During the COVID-19 Pandemic
A Systemic Lupus Erythematosus Incidence Surveillance Report Among DoD Beneficiaries During the COVID-19 Pandemic
- Izmirly PM, Ferucci ED, Somers EC, et al. Incidence rates of systemic lupus erythematosus in the USA: estimates from a meta-analysis of the Centers for Disease Control and Prevention national lupus registries. Lupus Sci Med. 2021;8(1):e000614. doi:10.1136/lupus-2021-000614
- Centers for Disease Control and Prevention. People with lupus. May 15, 2024. Accessed May 10, 2025. https:// www.cdc.gov/lupus/data-research/index.html
- Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526-1533. doi:10.1056/nejmoa021933
- Li ZX, Zeng S, Wu HX, Zhou Y. The risk of systemic lupus erythematosus associated with Epstein–Barr virus infection: a systematic review and meta-analysis. Clin Exp Med. 2019;19(1):23-36. doi:10.1007/s10238-018-0535-0
- Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4585. doi:10.1126/science.abd4585
- Chang SE, Feng A, Meng W, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. 2021;12(1):5417. doi:10.1038/s41467-021-25509-3
- Lee SJ, Yoon T, Ha JW, et al. Prevalence, clinical significance, and persistence of autoantibodies in COVID-19. Virol J. 2023;20(1):236. doi:10.1186/s12985-023-02191-z
- Woodruff MC, Ramonell RP, Haddad NS, et al. Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature. 2022;611(7934):139-147. doi:10.1038/s41586-022-05273-0
- Taeschler P, Cervia C, Zurbuchen Y, et al. Autoantibodies in COVID-19 correlate with antiviral humoral responses and distinct immune signatures. Allergy. 2022;77(8):2415-2430. doi:10.1111/all.15302
- Gracia-Ramos AE, Martin-Nares E, Hernández-Molina G. New onset of autoimmune diseases following COVID-19 diagnosis. Cells. 2021;10(12):3592 doi:10.3390/cells10123592
- Barnado A, Carroll R, Denny JC, Crofford L. Using IC-10-CM codes to identify patients with systemic lupus erythematosus in the electronic health record [abstract]. Arthritis Rheumatol. 2018;70(suppl 9):abstract 1692. Accessed May 10, 2025. https://acrabstracts.org/abstract/using-icd-10-cm-codes-to-identify-patients-with-systemic-lupus-erythematosus-in-the-electronic-health-record
- Feldman C, Curtis JR, Oates JC, Yazdany J, Izmirly P. Validating claims-based algorithms for a systemic lupus erythematosus diagnosis in Medicare data for informed use of the Lupus Index: a tool for geospatial research. Lupus Sci Med. 2024;11(2):e001329. doi:10.1136/lupus-2024-001329
- Moe SR, Haukeland H, Brunborg C, et al. POS1472: Accuracy of disease-specific ICD-10 code for incident systemic lupus erythematosus; results from a population-based cohort study set in Norway [abstract]. Ann Rheum Dis. 2023;82(suppl 1):1090-1091. doi:10.1136/annrheumdis-2023-eular.1189
- Denagamage P, Mabila SL, McQuistan AA. Trends and disparities in systemic lupus erythematosus incidence among U.S. active component service members, 2000–2022. MSMR. 2023;30(12):2-5.
- Chang R, Yen-Ting Chen T, Wang SI, Hung YM, Chen HY, Wei CJ. Risk of autoimmune diseases in patients with COVID-19: a retrospective cohort study. EClinicalMedicine. 2023;56:101783. doi:10.1016/j.eclinm.2022.101783
- Tesch F, Ehm F, Vivirito A, et al. Incident autoimmune diseases in association with SARS-CoV-2 infection: a matched cohort study. Clin Rheumatol. 2023;42(10):2905- 2914. doi:10.1007/s10067-023-06670-0
- Peng K, Li X, Yang D, et al. Risk of autoimmune diseases following COVID-19 and the potential protective effect from vaccination: a population-based cohort study. EClinicalMedicine. 2023;63:102154. doi:10.1016/j.eclinm.2023.102154
- US Department of Defense. Coronavirus: DOD response. Updated December 20, 2022. Accessed May 10, 2025. https://www.defense.gov/Spotlights/Coronavirus-DOD-Response/
- Elflein J. Number of cumulative cases of COVID-19 in the United States from January 20, 2020 to November 11, 2022, by week. Statista. https://www.statista.com/statistics/1103185/cumulative-coronavirus-covid19-cases-number-us-by-day
- Ye Z, Chen L, Zhong H, Cao L, Fu P, Xu J. Epidemiology and clinical characteristics of Epstein-Barr virus infection among children in Shanghai, China, 2017- 2022. Front Cell Infect Microbiol. 2023;13:1139068. doi:10.3389/fcimb.2023.1139068
- Johnson D, Jiang W. Infectious diseases, autoantibodies, and autoimmunity. J Autoimmun. 2023;137:102962. doi:10.1016/j.jaut.2022.102962
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
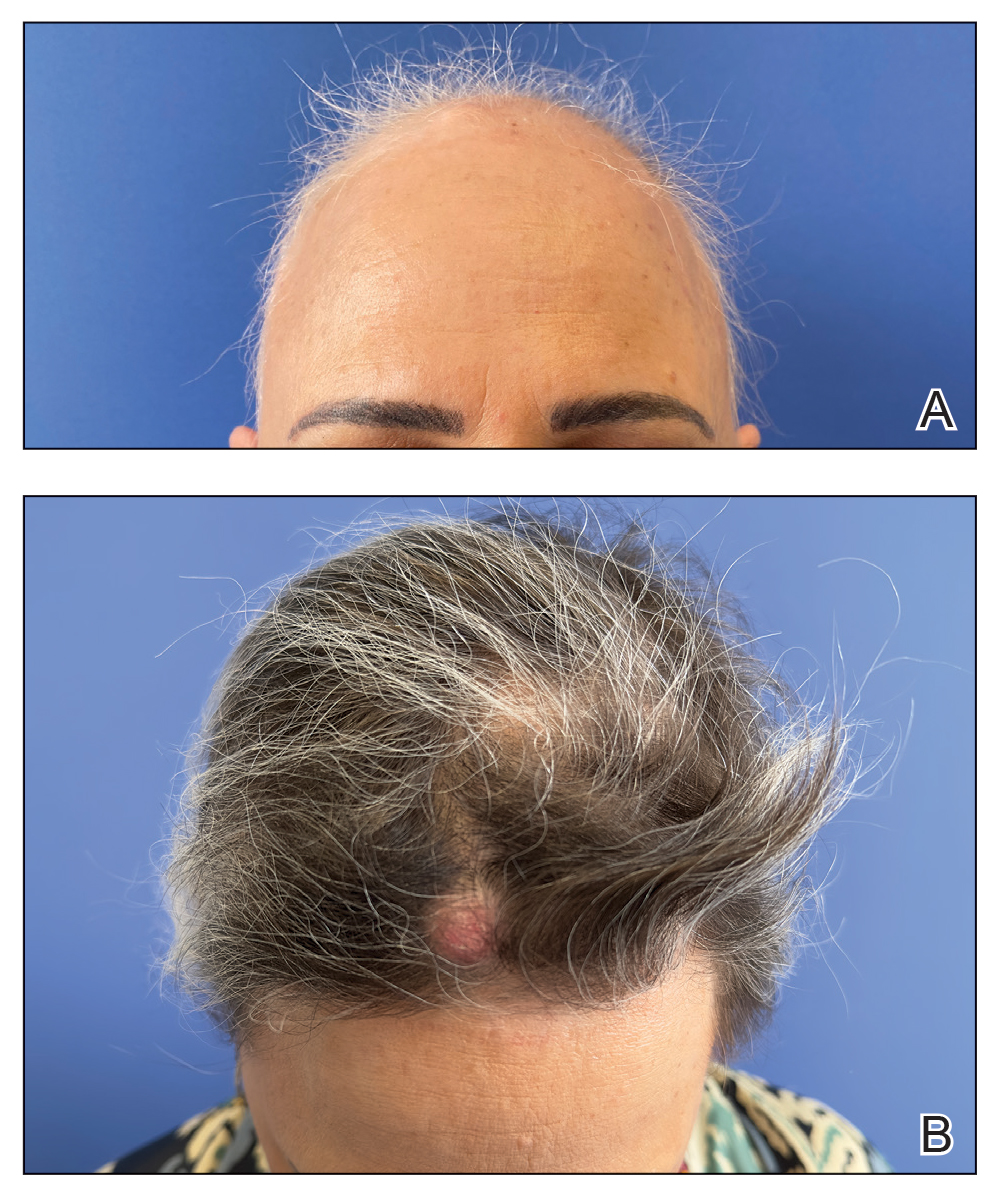
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
PRACTICE POINTS
- The rapid growth of trichilemmal cysts may serve as an indicator of a quick-responder phenotype to baricitinib in cases of alopecia areata (AA), although more evidence is needed.
- It is imperative to consider personal and family history of trichilemmal cysts prior to initiating baricitinib treatment for AA.

Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.

Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
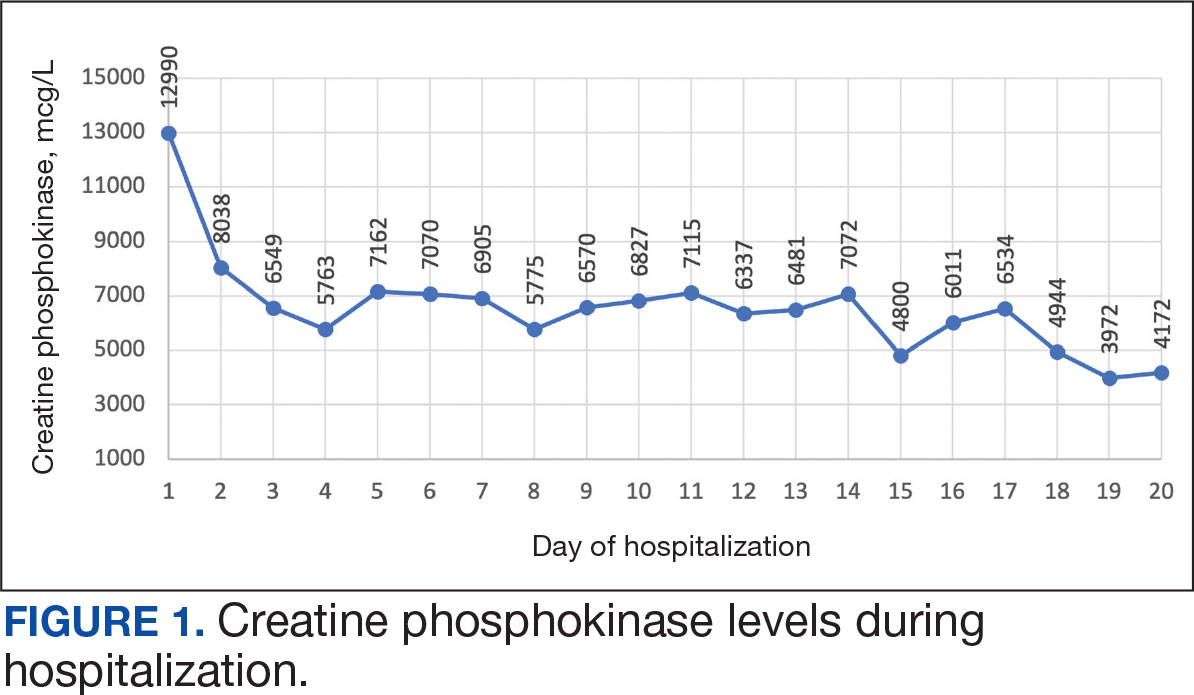
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
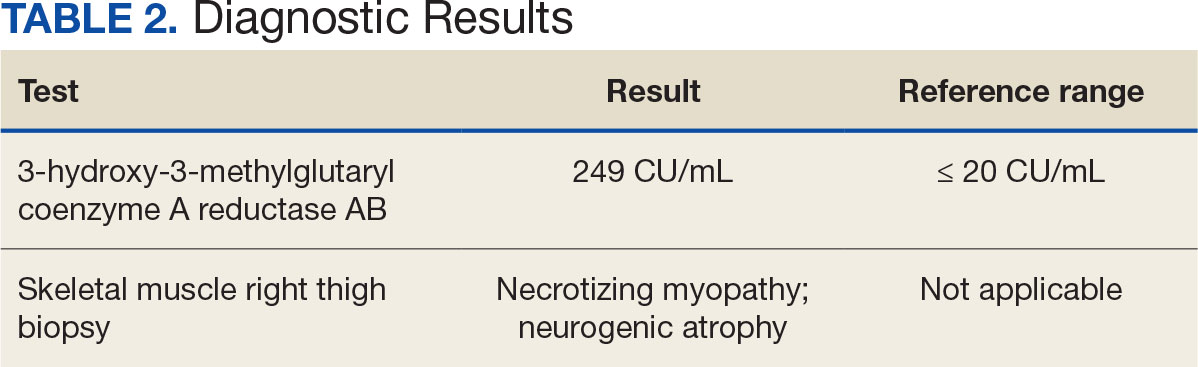
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
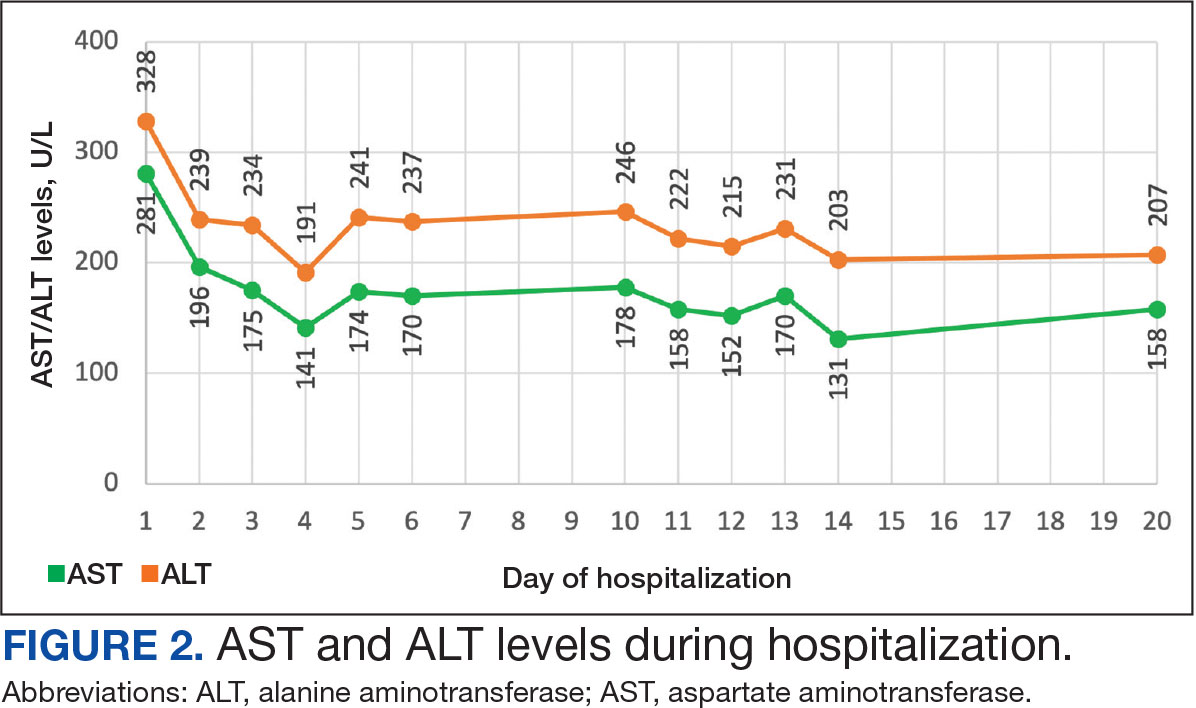
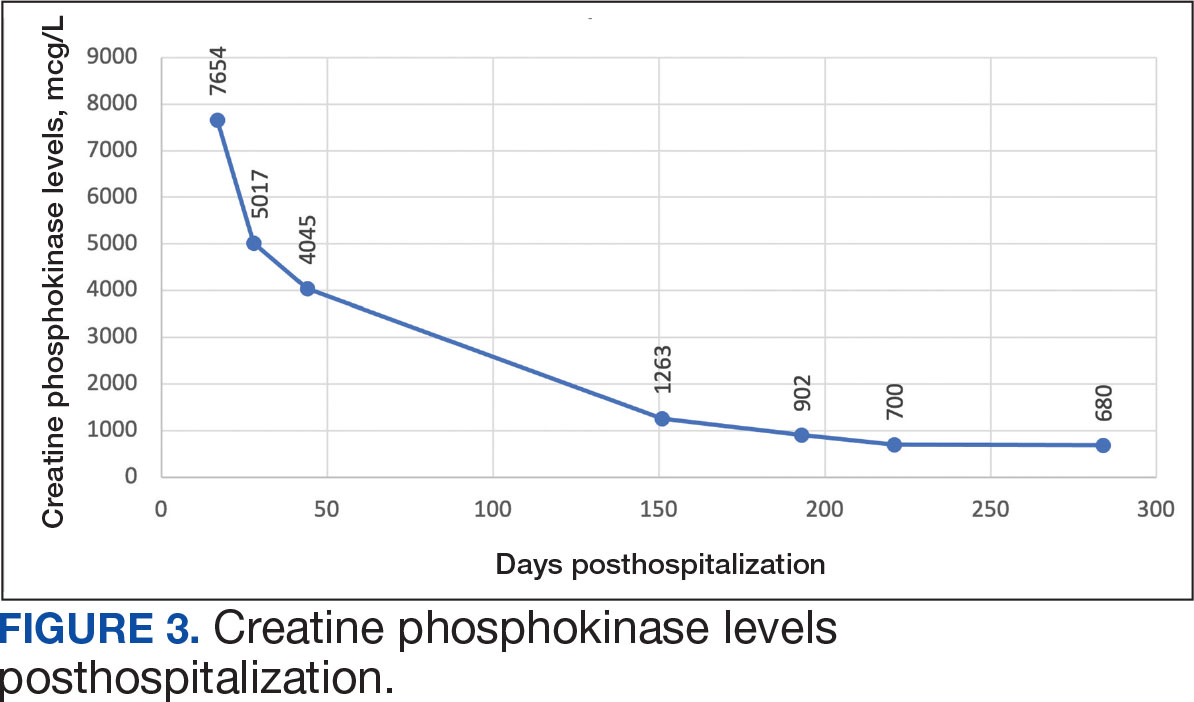
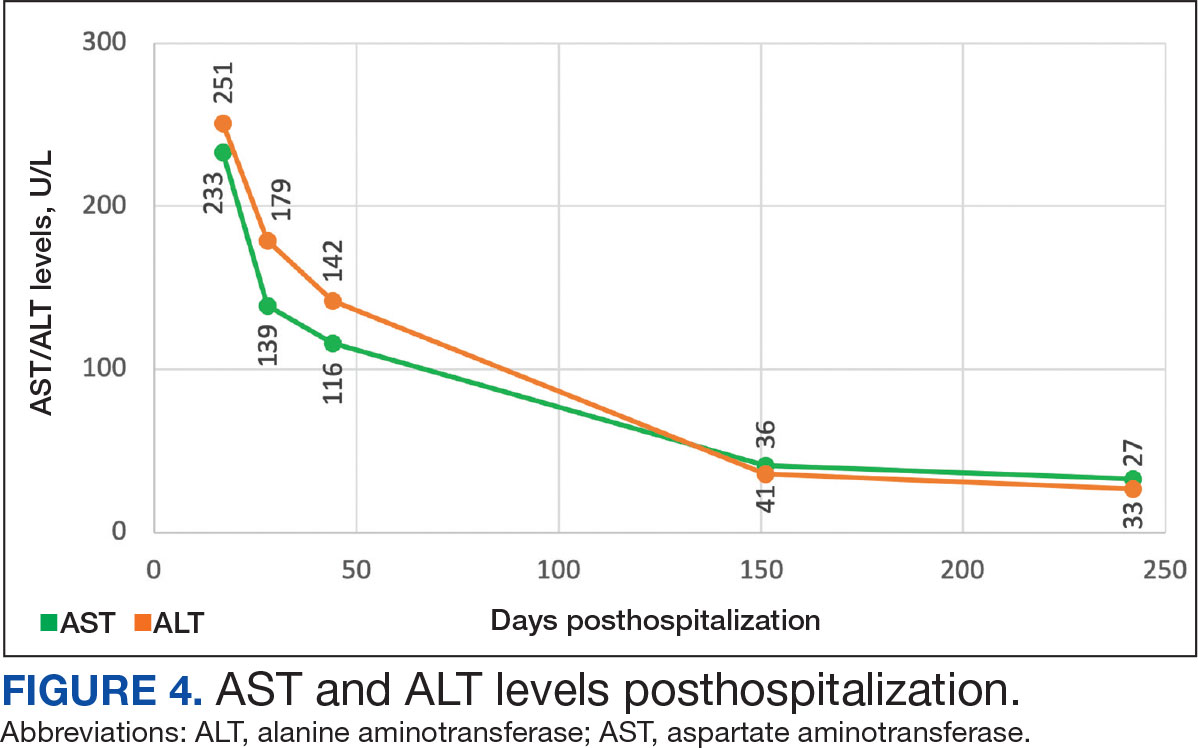
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.
Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.
Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
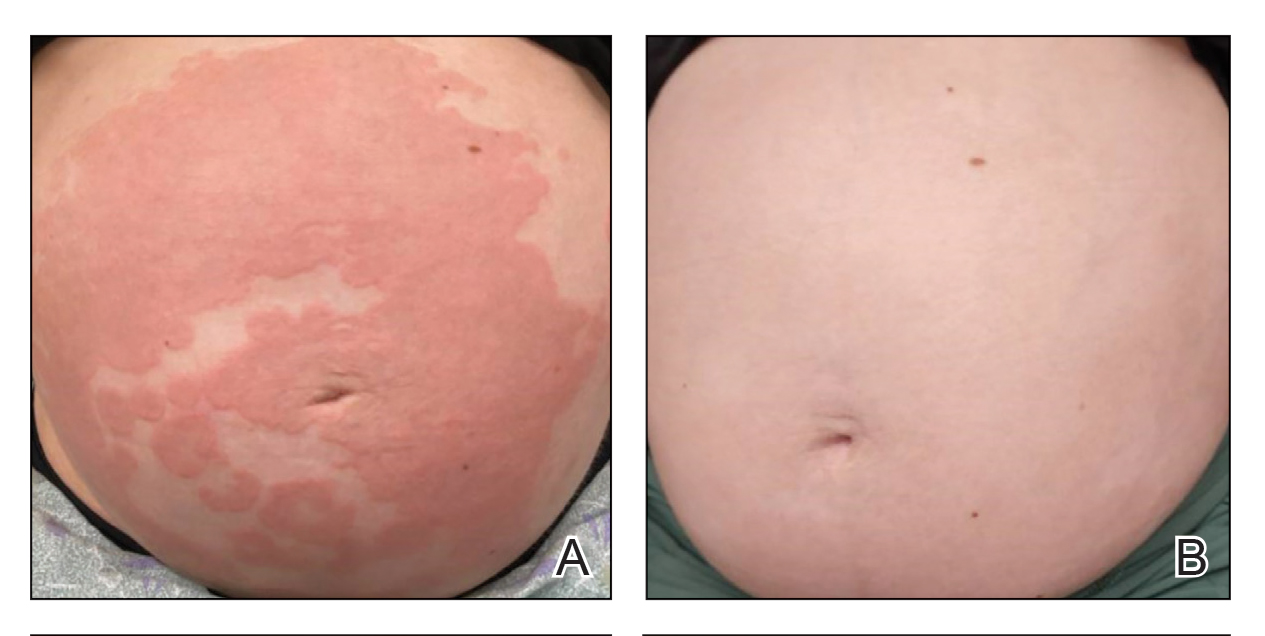
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
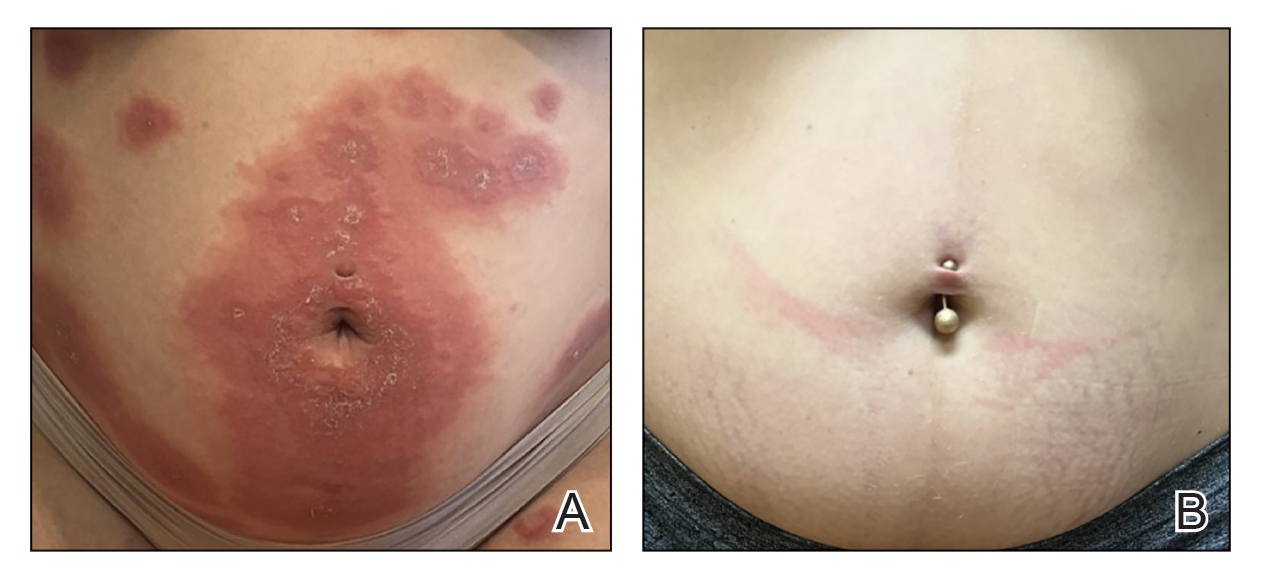
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
PRACTICE POINTS
- Dupilumab inhibits the IL-4Rα subunit, which is bound by IL‐4 and IL‐13, thereby reducing type 2 inflammation associated with pemphigoid gestationis (PG).
- Dupilumab may reduce the dose and duration of systemic corticosteroid therapy for PG, and its use in the second and third trimesters of pregnancy has been supported by emerging safety data.

Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
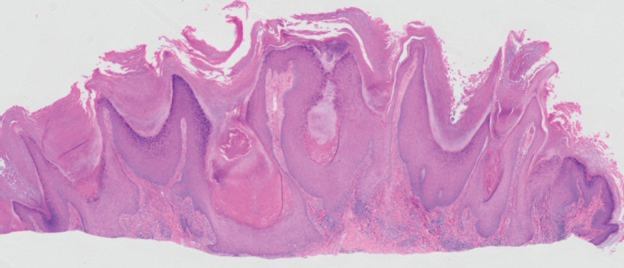
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
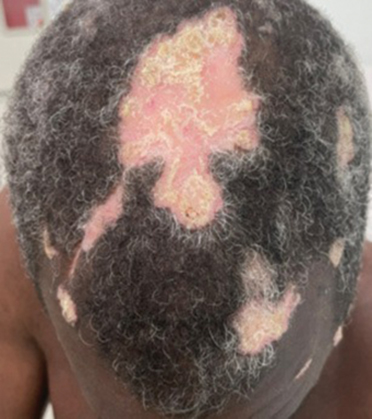
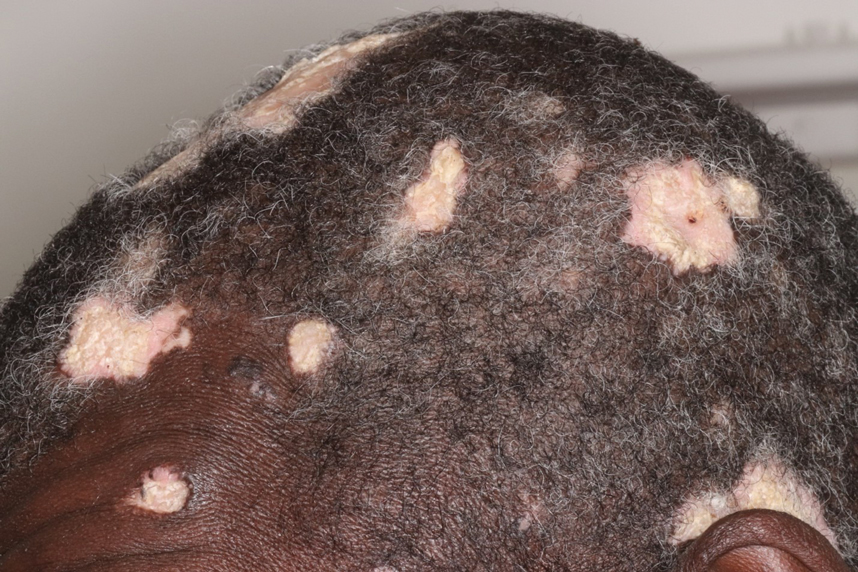
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
A 54-year-old man with no notable medical history presented to an outpatient dermatology clinic with multiple skin lesions on sun-exposed areas including the face, chest, scalp, and bilateral upper extremities. The patient reported that he had not seen a doctor for 26 years. He noted that the lesions had been present for many years but was unsure of the exact timeframe. Physical examination revealed verrucous plaques with a violaceous rim and central hypopigmentation on the chest, scalp, face, and arms. Scarring alopecia also was noted on the scalp with no associated pain or pruritus. Antinuclear antibody and extractable nuclear antigen tests were negative, and urine analysis was normal. A shave biopsy of the chest was performed for histopathologic evaluation. Rapid plasma reagin tests and HIV antibody tests also were performed.
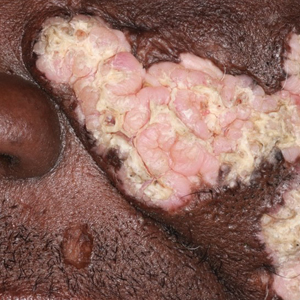
Biomarker Changes in Systemic Sclerosis–Associated Lung Disease Predict Therapy Response
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Around 5% of US Population Diagnosed With Autoimmune Disease
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.