User login
Nine VA Facilities to Open Research Trials for Psychedelics
Nine VA Facilities to Open Research Trials for Psychedelics
On Nov. 22, 2014, 8 years after he came back from Iraq with “crippling” posttraumatic stress disorder (PTSD), Jonathan Lubecky took his first dose of the psychedelic compound methylenedioxymethamphetamine (MDMA). Lubecky, a Marine, Army, and National Guard veteran, described his path to MDMA therapy in in the New Horizons in Health podcast.
After 5 suicide attempts and “the hundreds of times I thought about it or stood on a bridge or had a plan,” he felt he had run out of options. Then, in a counseling session, a psychiatric intern slid a piece of paper across the table to him. It read “Google MDMA PTSD.”
Luckily for Lubecky, a space in a clinical trial opened up, in which he had 8 hours of talk therapy with specially trained therapists, combined with MDMA. “MDMA is a tool that opens up the mind, body and spirit,” he said, “so you can heal and process all those memories and traumas that are causing yourissues. It puts you in a middle place where you can talk about trauma without having panic attacks, without your body betraying you, and look at it from a different perspective.” said he added, “It’s like doing therapy while being hugged by everyone who loves you in a bathtub full of puppies licking your face.” In 2023, 9 years after that first dose, Lubecky said, “I’ve been PTSD free longer than I had it.”
And now, in 2025, the research into psychedelic therapy for veterans like Lubecky is taking another step forward according to a report by Military.com. Nine VA facilities, in the Bronx, Los Angeles, Omaha, Palo Alto, Portland (Oregon), San Diego, San Francisco, West Haven, and White River Junction, are participating in long-term studies to test the safety and clinical impact of psychedelic compounds for PTSD, treatment-resistant depression, and anxiety disorders.
Early trials from Johns Hopkins University, the Multidisciplinary Association for Psychedelic Studies (MAPS), and others found significant symptom reductions for some participants with chronic PTSD. MAPP2, the multisite phase 3 study that extended the findings of MAPP1, found that MDMA-assisted therapy significantly improved PTSD symptoms and functional impairment, compared with placebo-assisted therapy. Notably, of the 52 participants (including 16 veterans) 45 (86%) achieved a clinically meaningful benefit, and 37 (71%) no longer met criteria for PTSD by study end. Despite the promising findings, a US Food and Drug Administration (FDA) advisory panel recommended against approving the treatment.
In 2024 the VA issued a request for applications for proposals from its network of VA researchers and academic institutions to gather “definitive scientific evidence” on the potential efficacy and safety of psychedelic compounds, such as MDMA and psilocybin, when used in conjunction with psychotherapy. It would be the first time since the 1960s that the VA had funded research on such compounds.
Funding proposals for such research have cycled in and out of Congress for years, but have gathered more steam in the last few years. The 2024 National Defense Authorization Act directed the US Department of Defense to establish a process for funding clinical research into the use of certain psychedelic substances to treat PTSD and traumatic brain injury. In April 2024, Representatives Lou Correa (D-CA) and Jack Bergman (R-MI), cochairs of the Psychedelics Advancing Therapies (PATH) caucus, introduced the Innovative Therapies Centers of Excellence Act of 2025, bipartisan legislation that would increase federally funded research on innovative therapies to treat veterans with PTSD, substance use disorder, and depression. It would also, if enacted, direct the VA to create ≥ 5 dedicated centers of excellence to study the therapeutic uses of psychedelic substances. The bill has also been endorsed by the American Legion, Veterans of Foreign Wars, Iraq and Afghanistan Veterans of America, Disabled American Veterans, and the Wounded Warrior Project.
The current administration has two strong high-level supporters of psychedelics research: VA Secretary Doug Collins and US Department of Health and Human Service Secretary Robert F. Kennedy Jr. Sec. Kennedy has castigated the FDA for what he calls “aggressive suppression” of alternative and complementary treatments, including psychedelics. This, although the FDA granted breakthrough therapy status for MDMA for treating PTSD and psilocybin for treating depression in 2018 and 2019, respectively, as well a pivotal draft guidance in 2023 for the development of psychedelic drugs for psychiatric disorders, substance use disorders, and various medical conditions.
Collins, citing an “eye-opening” discussion with Kennedy, enthusiastically backs the research into psychedelics. In a May 2025 hearing that was mainly a series of testy exchanges about his proposed budget slashing, he emphasized the importance of keeping and expanding VA programs and studies on psychedelic treatments, something he has been advocating for since the beginning of his appointment. “We want to make sure we’re not closing off any outlet for a veteran who could be helped by these programs,” he said.
Taking the intern’s advice to look into MDMA, Jonathan Lubecky said, was one of the best decisions he’d ever made. But “it’s not the MDMA that fixes you,” he said. “It’s the therapy. It’s the therapist working with you and you doing the hard work.”
On Nov. 22, 2014, 8 years after he came back from Iraq with “crippling” posttraumatic stress disorder (PTSD), Jonathan Lubecky took his first dose of the psychedelic compound methylenedioxymethamphetamine (MDMA). Lubecky, a Marine, Army, and National Guard veteran, described his path to MDMA therapy in in the New Horizons in Health podcast.
After 5 suicide attempts and “the hundreds of times I thought about it or stood on a bridge or had a plan,” he felt he had run out of options. Then, in a counseling session, a psychiatric intern slid a piece of paper across the table to him. It read “Google MDMA PTSD.”
Luckily for Lubecky, a space in a clinical trial opened up, in which he had 8 hours of talk therapy with specially trained therapists, combined with MDMA. “MDMA is a tool that opens up the mind, body and spirit,” he said, “so you can heal and process all those memories and traumas that are causing yourissues. It puts you in a middle place where you can talk about trauma without having panic attacks, without your body betraying you, and look at it from a different perspective.” said he added, “It’s like doing therapy while being hugged by everyone who loves you in a bathtub full of puppies licking your face.” In 2023, 9 years after that first dose, Lubecky said, “I’ve been PTSD free longer than I had it.”
And now, in 2025, the research into psychedelic therapy for veterans like Lubecky is taking another step forward according to a report by Military.com. Nine VA facilities, in the Bronx, Los Angeles, Omaha, Palo Alto, Portland (Oregon), San Diego, San Francisco, West Haven, and White River Junction, are participating in long-term studies to test the safety and clinical impact of psychedelic compounds for PTSD, treatment-resistant depression, and anxiety disorders.
Early trials from Johns Hopkins University, the Multidisciplinary Association for Psychedelic Studies (MAPS), and others found significant symptom reductions for some participants with chronic PTSD. MAPP2, the multisite phase 3 study that extended the findings of MAPP1, found that MDMA-assisted therapy significantly improved PTSD symptoms and functional impairment, compared with placebo-assisted therapy. Notably, of the 52 participants (including 16 veterans) 45 (86%) achieved a clinically meaningful benefit, and 37 (71%) no longer met criteria for PTSD by study end. Despite the promising findings, a US Food and Drug Administration (FDA) advisory panel recommended against approving the treatment.
In 2024 the VA issued a request for applications for proposals from its network of VA researchers and academic institutions to gather “definitive scientific evidence” on the potential efficacy and safety of psychedelic compounds, such as MDMA and psilocybin, when used in conjunction with psychotherapy. It would be the first time since the 1960s that the VA had funded research on such compounds.
Funding proposals for such research have cycled in and out of Congress for years, but have gathered more steam in the last few years. The 2024 National Defense Authorization Act directed the US Department of Defense to establish a process for funding clinical research into the use of certain psychedelic substances to treat PTSD and traumatic brain injury. In April 2024, Representatives Lou Correa (D-CA) and Jack Bergman (R-MI), cochairs of the Psychedelics Advancing Therapies (PATH) caucus, introduced the Innovative Therapies Centers of Excellence Act of 2025, bipartisan legislation that would increase federally funded research on innovative therapies to treat veterans with PTSD, substance use disorder, and depression. It would also, if enacted, direct the VA to create ≥ 5 dedicated centers of excellence to study the therapeutic uses of psychedelic substances. The bill has also been endorsed by the American Legion, Veterans of Foreign Wars, Iraq and Afghanistan Veterans of America, Disabled American Veterans, and the Wounded Warrior Project.
The current administration has two strong high-level supporters of psychedelics research: VA Secretary Doug Collins and US Department of Health and Human Service Secretary Robert F. Kennedy Jr. Sec. Kennedy has castigated the FDA for what he calls “aggressive suppression” of alternative and complementary treatments, including psychedelics. This, although the FDA granted breakthrough therapy status for MDMA for treating PTSD and psilocybin for treating depression in 2018 and 2019, respectively, as well a pivotal draft guidance in 2023 for the development of psychedelic drugs for psychiatric disorders, substance use disorders, and various medical conditions.
Collins, citing an “eye-opening” discussion with Kennedy, enthusiastically backs the research into psychedelics. In a May 2025 hearing that was mainly a series of testy exchanges about his proposed budget slashing, he emphasized the importance of keeping and expanding VA programs and studies on psychedelic treatments, something he has been advocating for since the beginning of his appointment. “We want to make sure we’re not closing off any outlet for a veteran who could be helped by these programs,” he said.
Taking the intern’s advice to look into MDMA, Jonathan Lubecky said, was one of the best decisions he’d ever made. But “it’s not the MDMA that fixes you,” he said. “It’s the therapy. It’s the therapist working with you and you doing the hard work.”
On Nov. 22, 2014, 8 years after he came back from Iraq with “crippling” posttraumatic stress disorder (PTSD), Jonathan Lubecky took his first dose of the psychedelic compound methylenedioxymethamphetamine (MDMA). Lubecky, a Marine, Army, and National Guard veteran, described his path to MDMA therapy in in the New Horizons in Health podcast.
After 5 suicide attempts and “the hundreds of times I thought about it or stood on a bridge or had a plan,” he felt he had run out of options. Then, in a counseling session, a psychiatric intern slid a piece of paper across the table to him. It read “Google MDMA PTSD.”
Luckily for Lubecky, a space in a clinical trial opened up, in which he had 8 hours of talk therapy with specially trained therapists, combined with MDMA. “MDMA is a tool that opens up the mind, body and spirit,” he said, “so you can heal and process all those memories and traumas that are causing yourissues. It puts you in a middle place where you can talk about trauma without having panic attacks, without your body betraying you, and look at it from a different perspective.” said he added, “It’s like doing therapy while being hugged by everyone who loves you in a bathtub full of puppies licking your face.” In 2023, 9 years after that first dose, Lubecky said, “I’ve been PTSD free longer than I had it.”
And now, in 2025, the research into psychedelic therapy for veterans like Lubecky is taking another step forward according to a report by Military.com. Nine VA facilities, in the Bronx, Los Angeles, Omaha, Palo Alto, Portland (Oregon), San Diego, San Francisco, West Haven, and White River Junction, are participating in long-term studies to test the safety and clinical impact of psychedelic compounds for PTSD, treatment-resistant depression, and anxiety disorders.
Early trials from Johns Hopkins University, the Multidisciplinary Association for Psychedelic Studies (MAPS), and others found significant symptom reductions for some participants with chronic PTSD. MAPP2, the multisite phase 3 study that extended the findings of MAPP1, found that MDMA-assisted therapy significantly improved PTSD symptoms and functional impairment, compared with placebo-assisted therapy. Notably, of the 52 participants (including 16 veterans) 45 (86%) achieved a clinically meaningful benefit, and 37 (71%) no longer met criteria for PTSD by study end. Despite the promising findings, a US Food and Drug Administration (FDA) advisory panel recommended against approving the treatment.
In 2024 the VA issued a request for applications for proposals from its network of VA researchers and academic institutions to gather “definitive scientific evidence” on the potential efficacy and safety of psychedelic compounds, such as MDMA and psilocybin, when used in conjunction with psychotherapy. It would be the first time since the 1960s that the VA had funded research on such compounds.
Funding proposals for such research have cycled in and out of Congress for years, but have gathered more steam in the last few years. The 2024 National Defense Authorization Act directed the US Department of Defense to establish a process for funding clinical research into the use of certain psychedelic substances to treat PTSD and traumatic brain injury. In April 2024, Representatives Lou Correa (D-CA) and Jack Bergman (R-MI), cochairs of the Psychedelics Advancing Therapies (PATH) caucus, introduced the Innovative Therapies Centers of Excellence Act of 2025, bipartisan legislation that would increase federally funded research on innovative therapies to treat veterans with PTSD, substance use disorder, and depression. It would also, if enacted, direct the VA to create ≥ 5 dedicated centers of excellence to study the therapeutic uses of psychedelic substances. The bill has also been endorsed by the American Legion, Veterans of Foreign Wars, Iraq and Afghanistan Veterans of America, Disabled American Veterans, and the Wounded Warrior Project.
The current administration has two strong high-level supporters of psychedelics research: VA Secretary Doug Collins and US Department of Health and Human Service Secretary Robert F. Kennedy Jr. Sec. Kennedy has castigated the FDA for what he calls “aggressive suppression” of alternative and complementary treatments, including psychedelics. This, although the FDA granted breakthrough therapy status for MDMA for treating PTSD and psilocybin for treating depression in 2018 and 2019, respectively, as well a pivotal draft guidance in 2023 for the development of psychedelic drugs for psychiatric disorders, substance use disorders, and various medical conditions.
Collins, citing an “eye-opening” discussion with Kennedy, enthusiastically backs the research into psychedelics. In a May 2025 hearing that was mainly a series of testy exchanges about his proposed budget slashing, he emphasized the importance of keeping and expanding VA programs and studies on psychedelic treatments, something he has been advocating for since the beginning of his appointment. “We want to make sure we’re not closing off any outlet for a veteran who could be helped by these programs,” he said.
Taking the intern’s advice to look into MDMA, Jonathan Lubecky said, was one of the best decisions he’d ever made. But “it’s not the MDMA that fixes you,” he said. “It’s the therapy. It’s the therapist working with you and you doing the hard work.”
Nine VA Facilities to Open Research Trials for Psychedelics
Nine VA Facilities to Open Research Trials for Psychedelics
New Drug Eases Side Effects of Weight-Loss Meds
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
FROM OBESITY WEEK 2025

Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
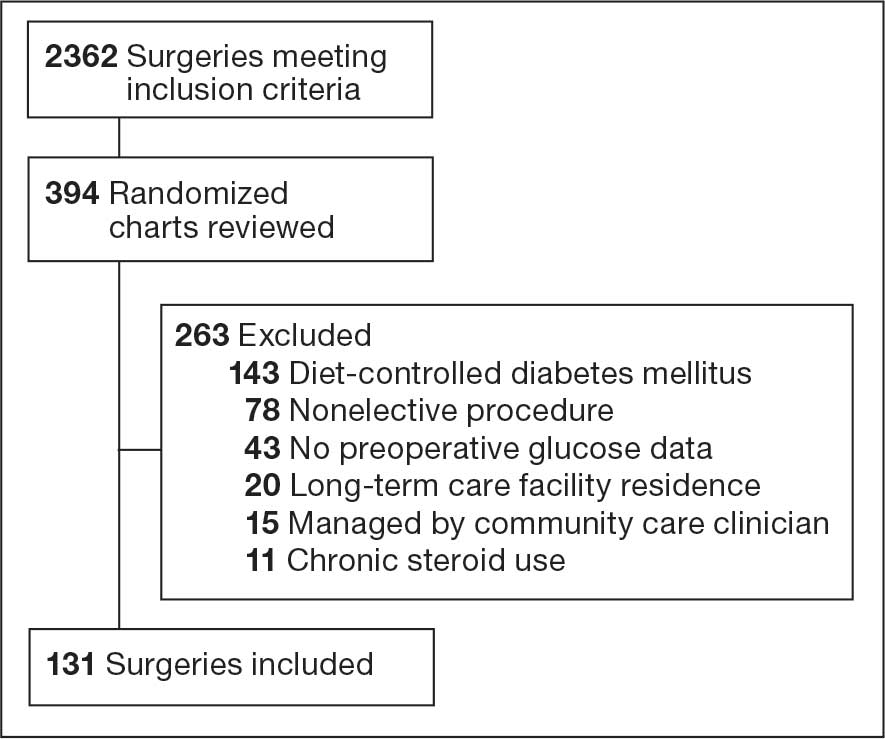
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
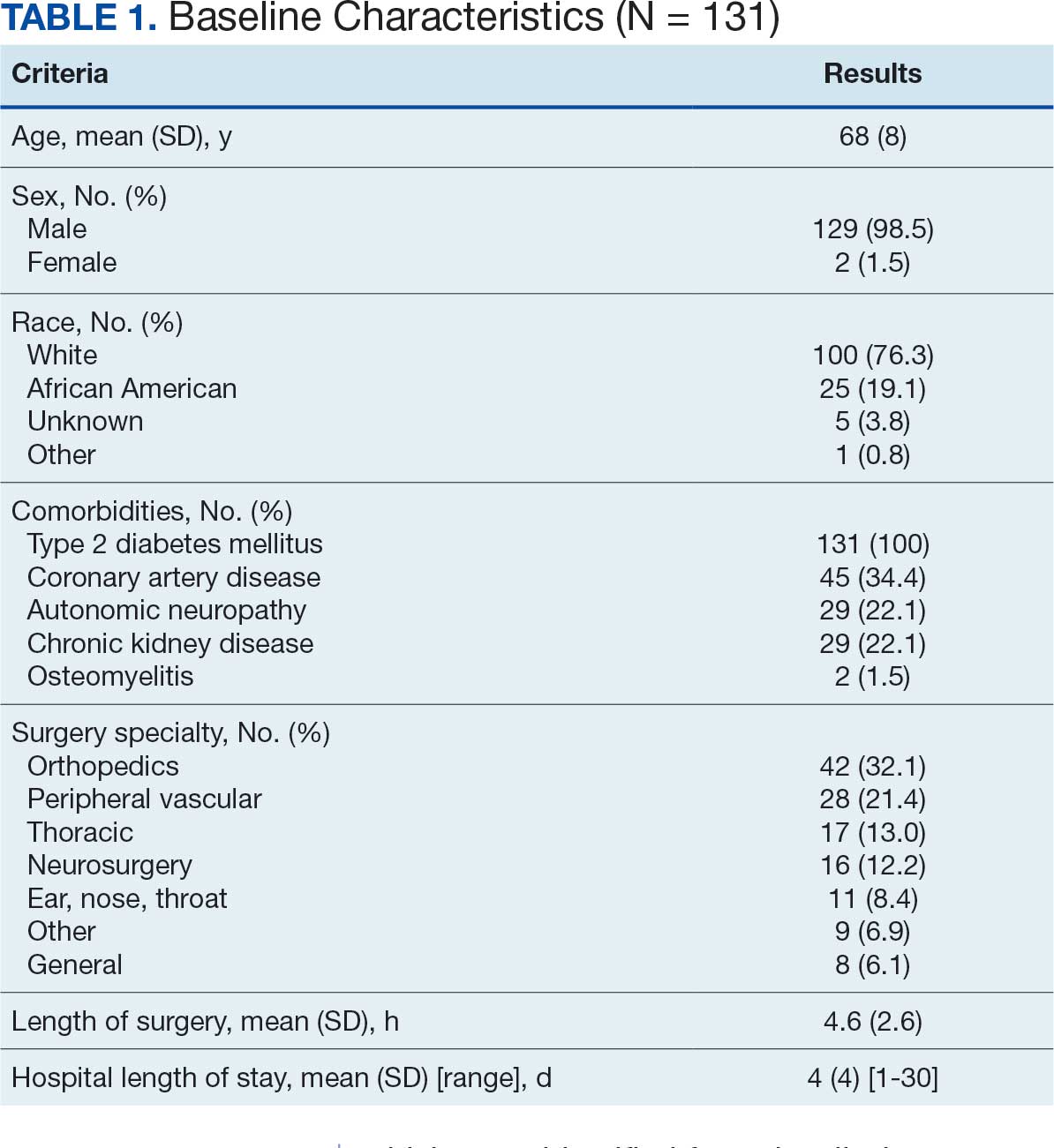
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
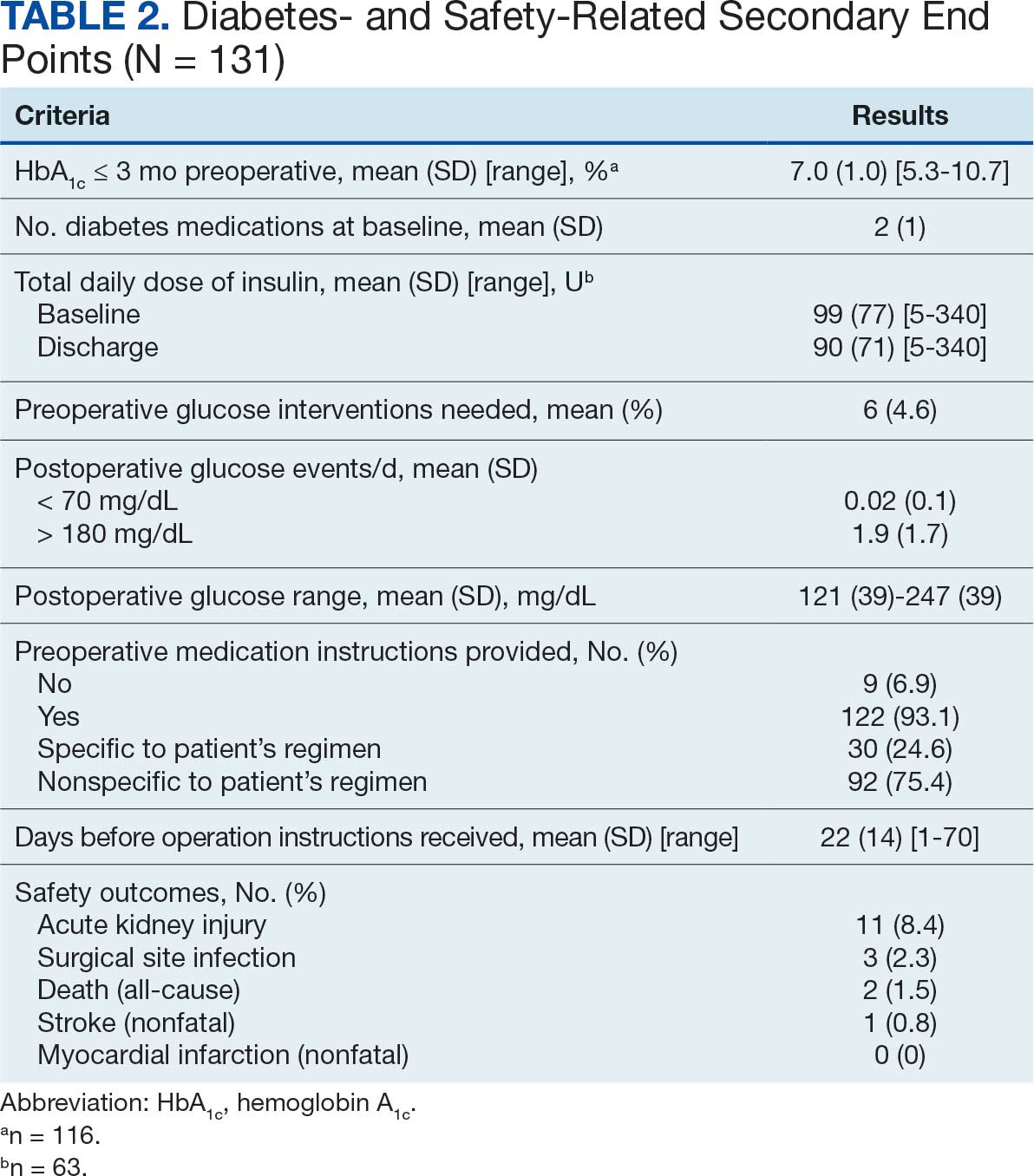
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
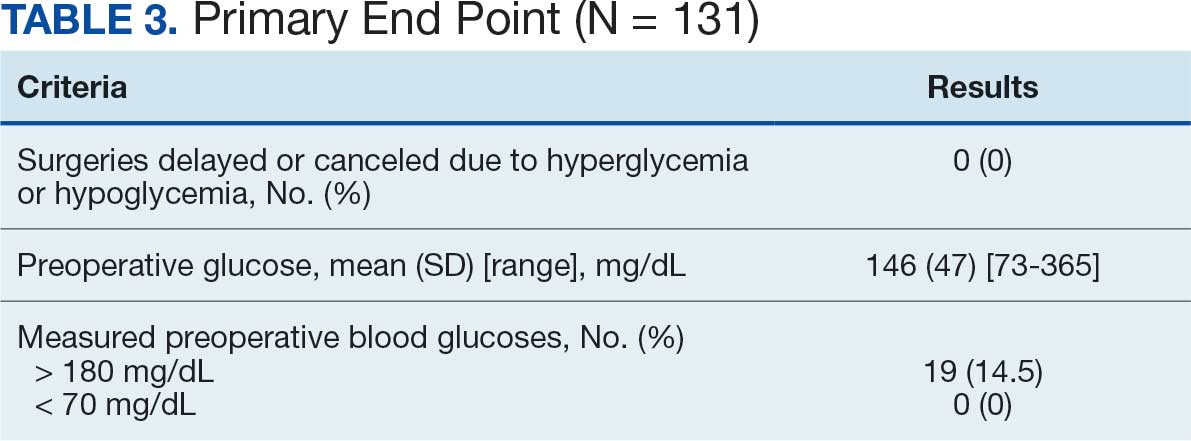
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).

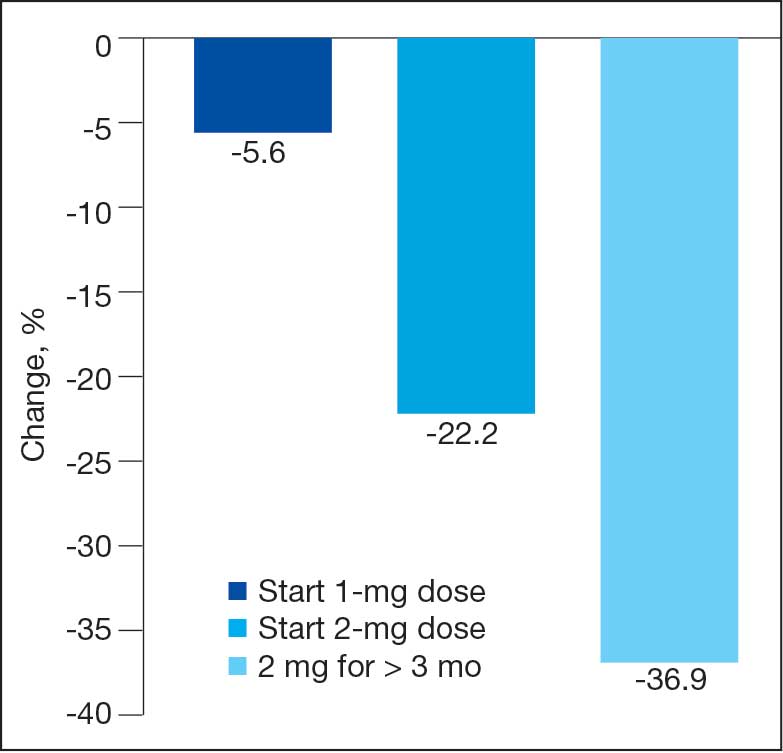
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
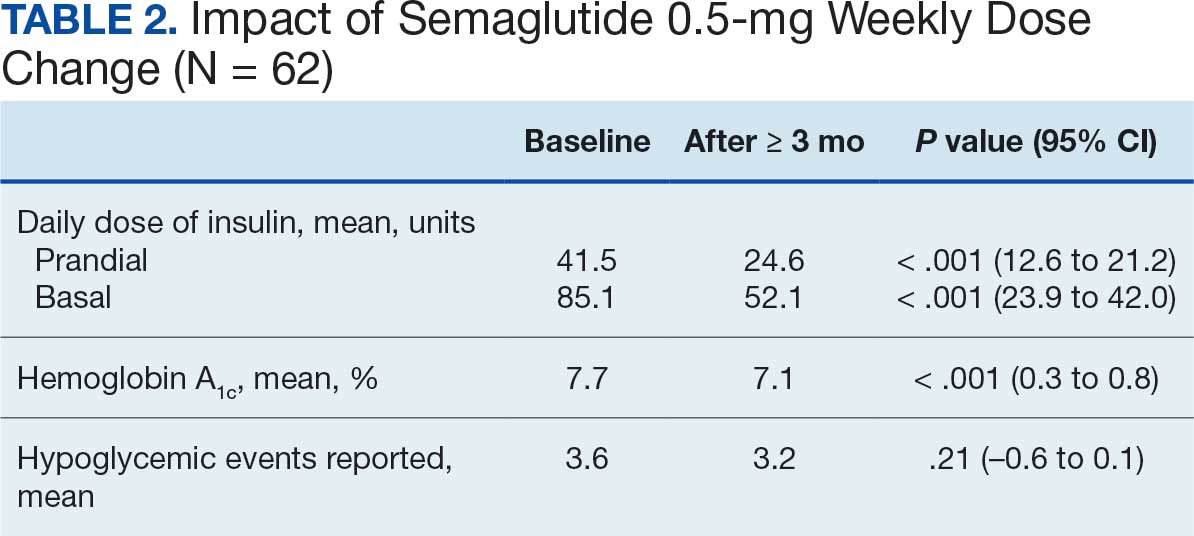
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
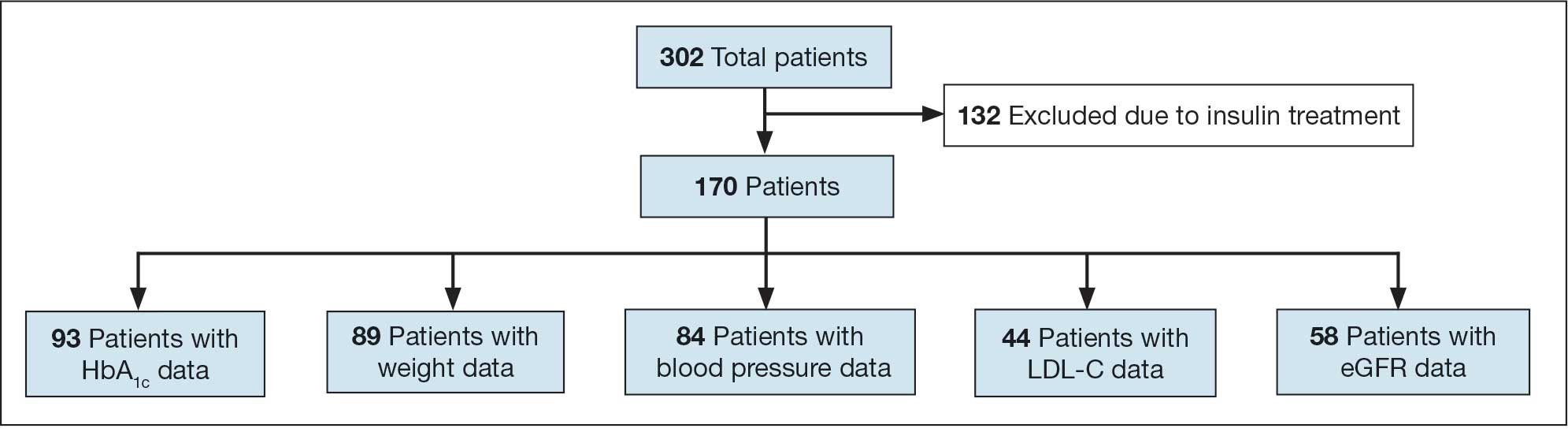
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
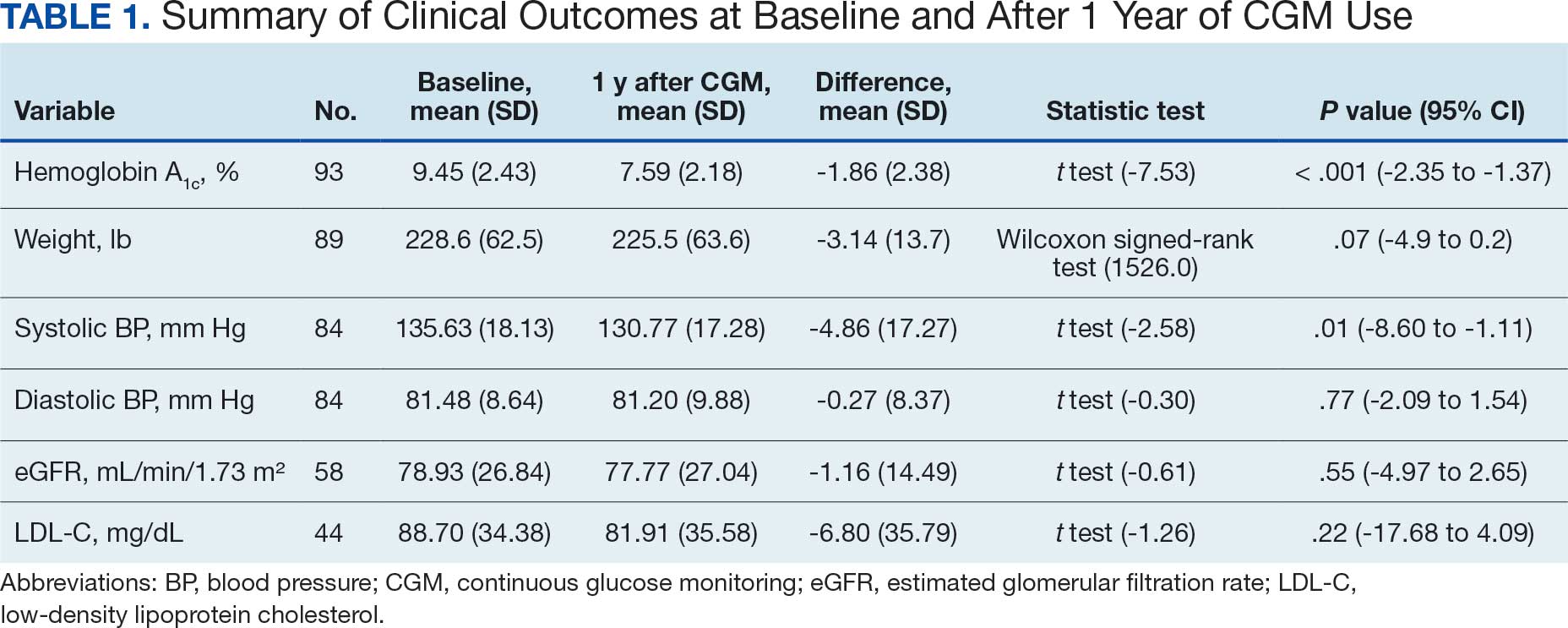
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
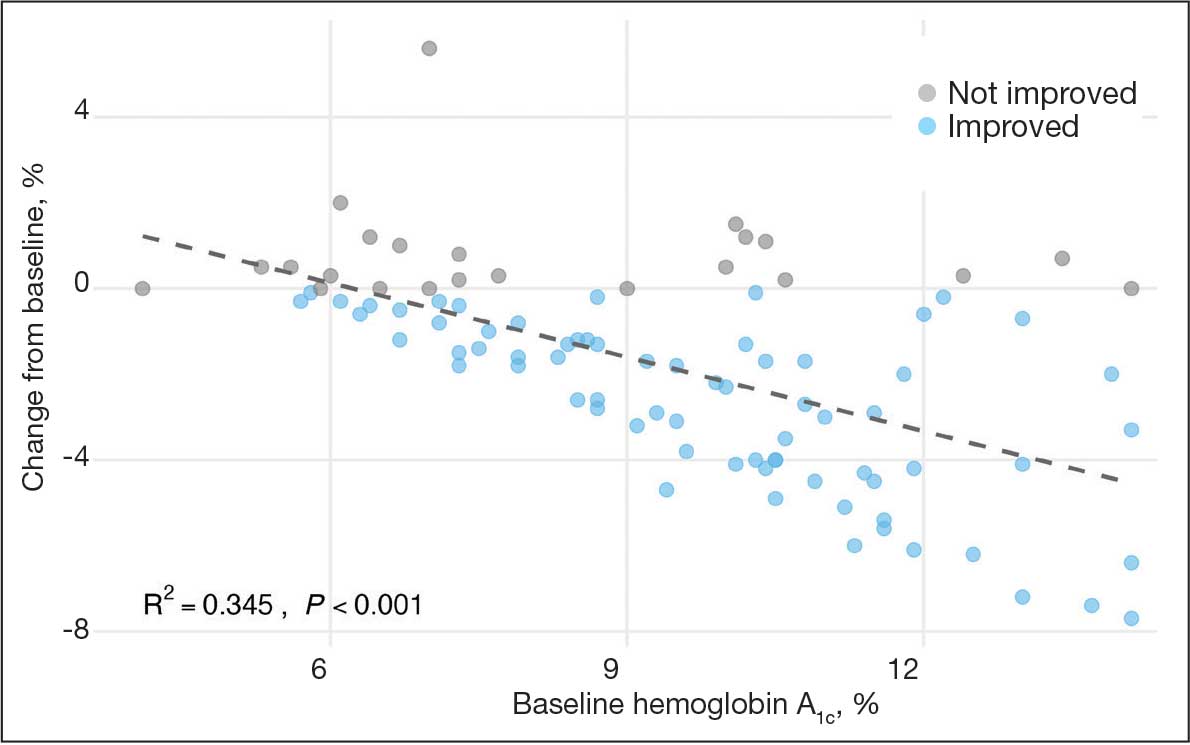
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
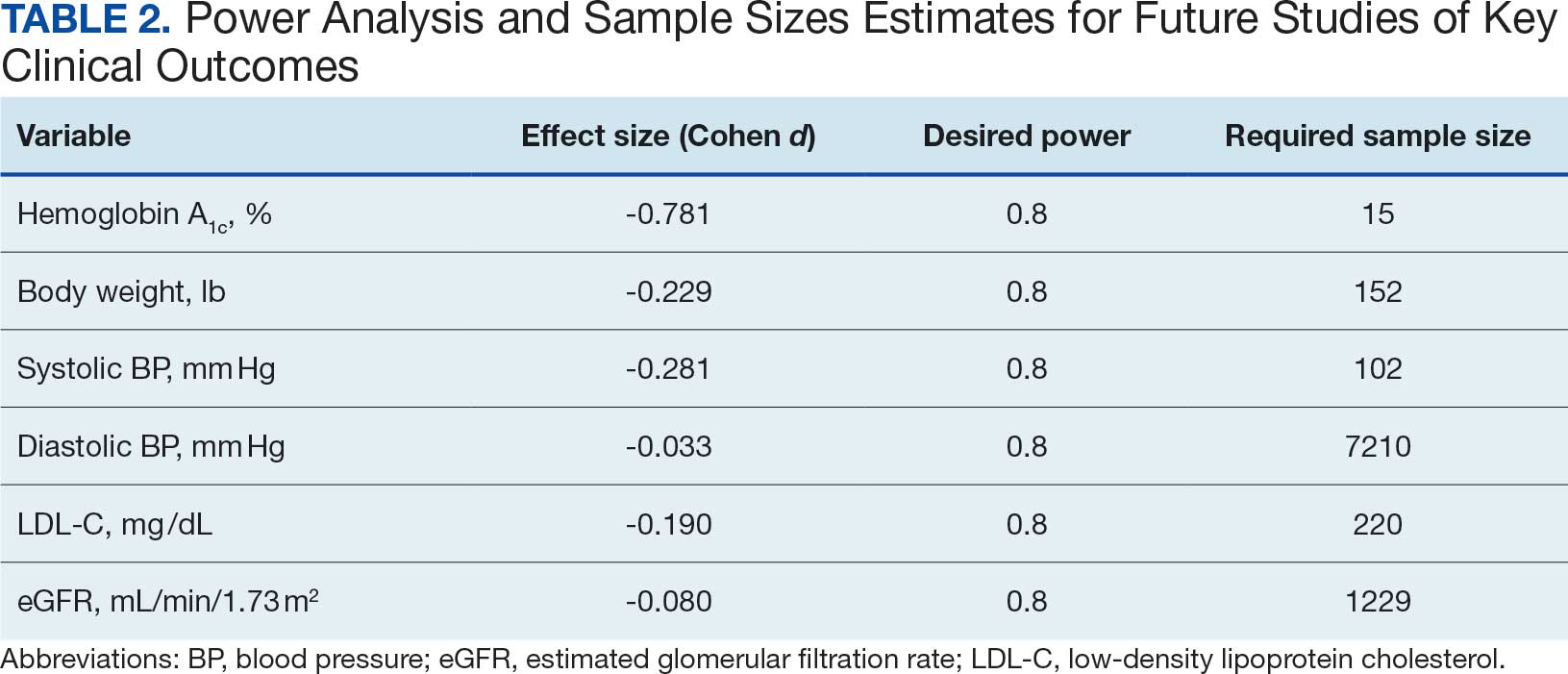
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
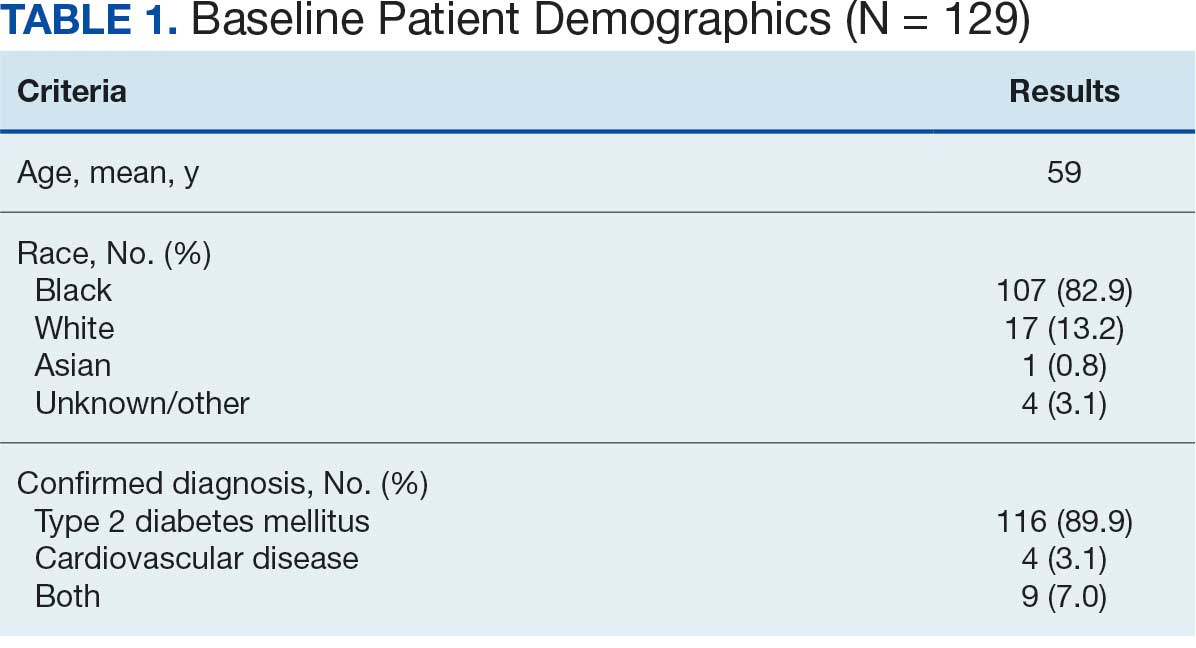
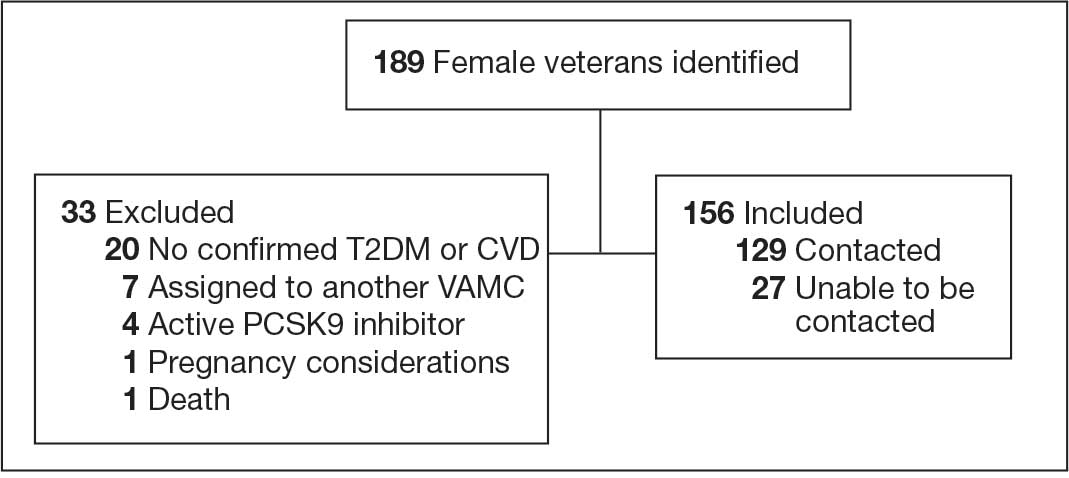
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).

From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
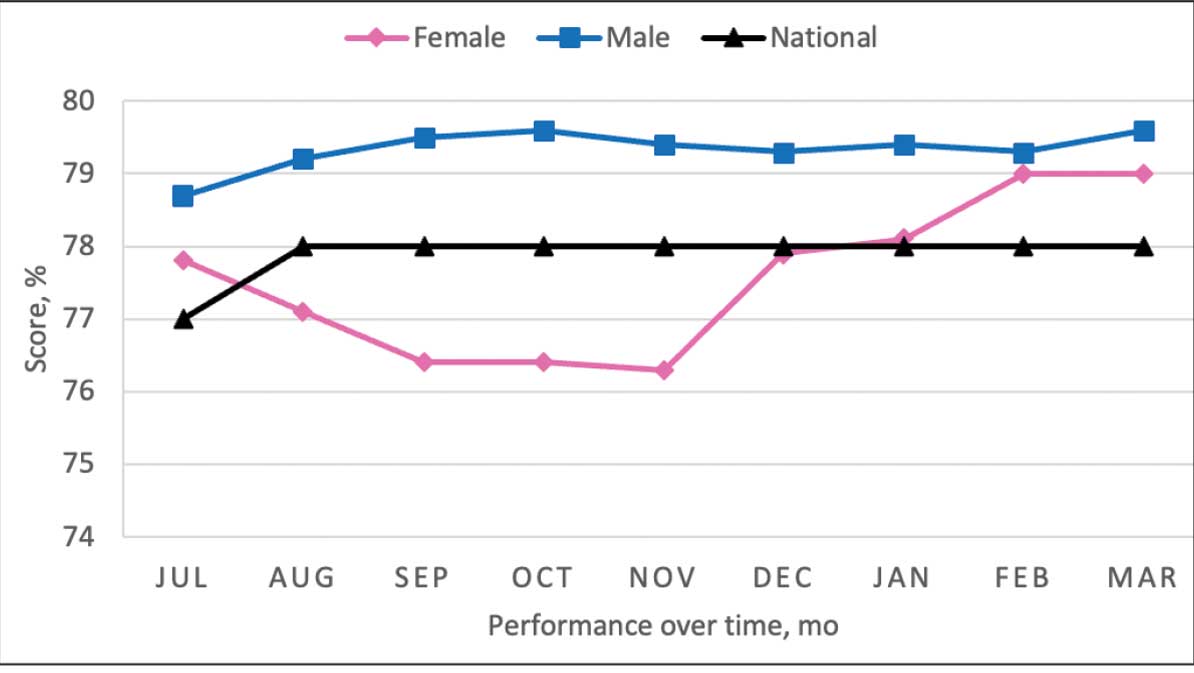
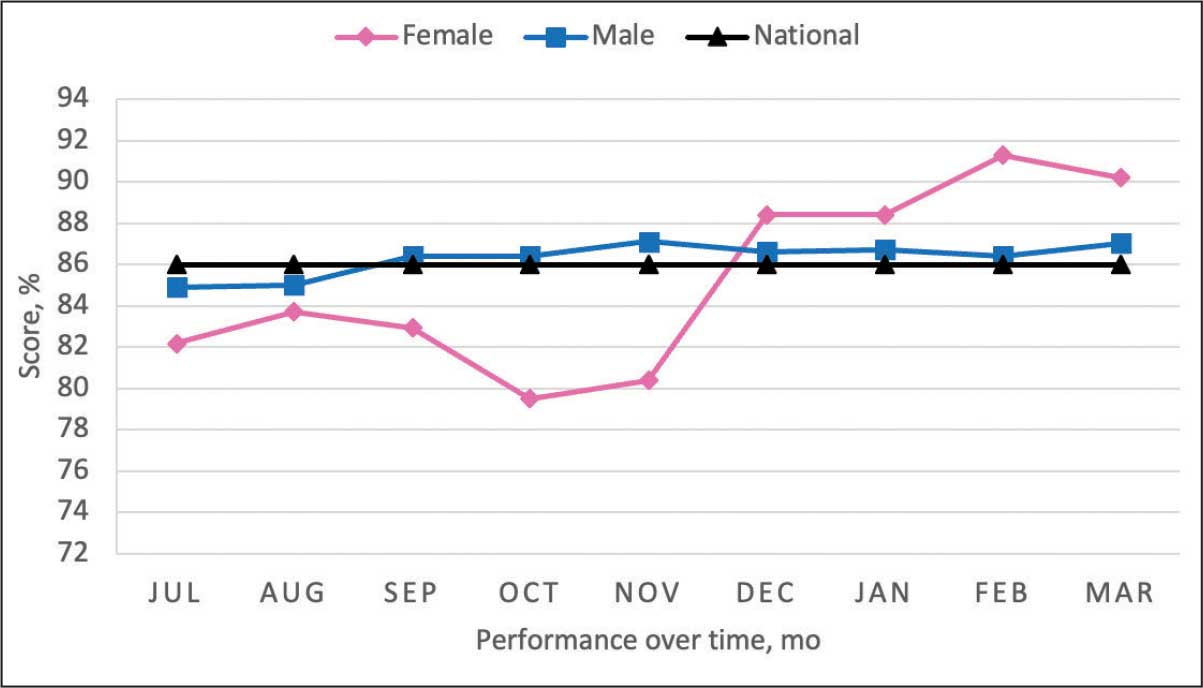
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
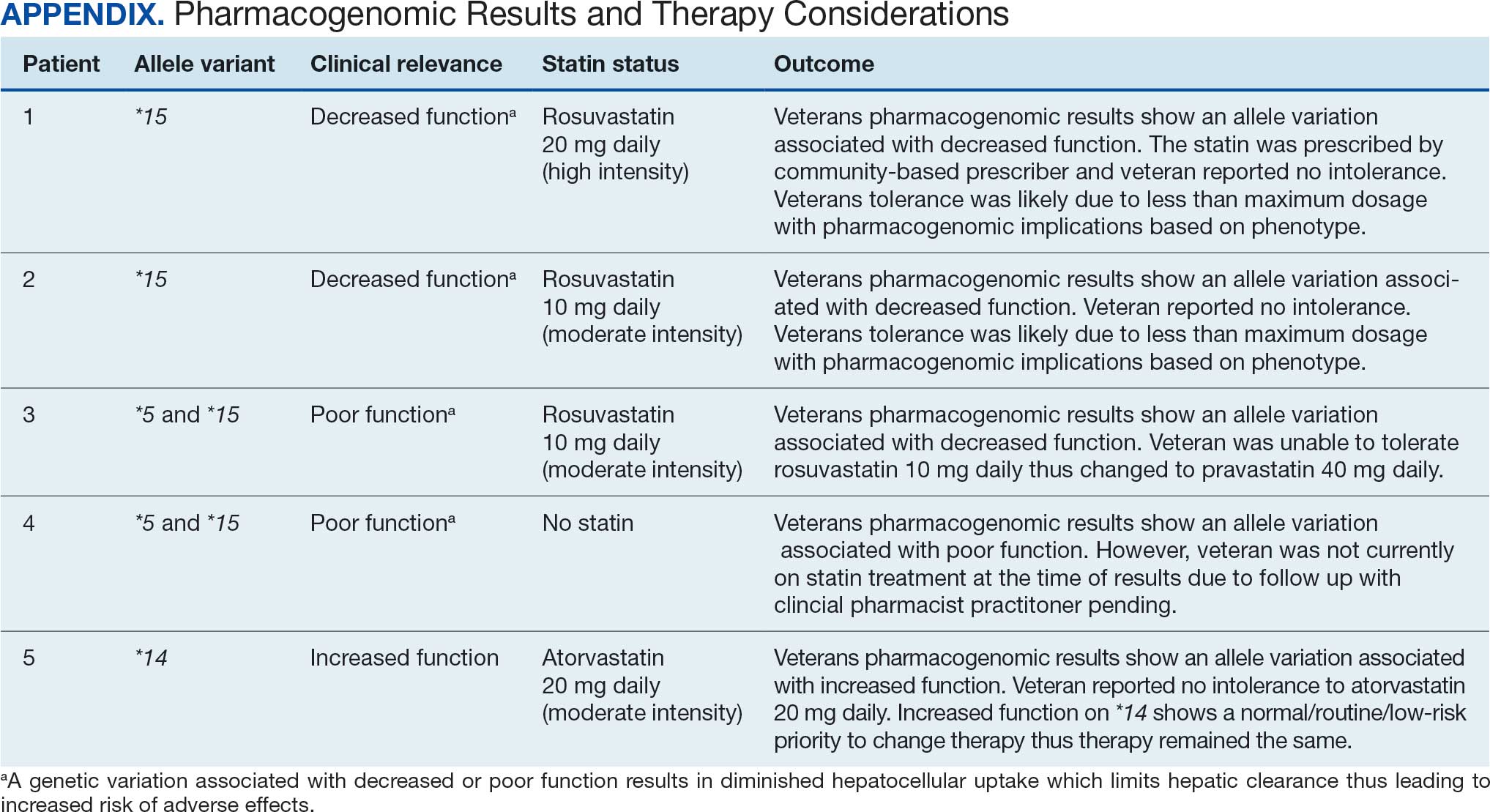
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).
From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).
From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Evaluating the Implementation of 60-minute Iron Dextran Infusions at a Rural Health Center
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.
Implementation of a VHA Virtual Oncology Training Pilot Program for Clinical Pharmacists
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Daratumumab and Darbepoetin for Refractory Warm Autoimmune Hemolytic Anemia: A Novel Duo for a Tough Case
Background
Warm autoimmune hemolytic anemia (wAIHA) is traditionally treated with immunosuppresimmunosuppression, and management of refractory disease is often a challenge. The anti-CD38 antibody daratumumab is emerging as a promising treatment for refractory wAIHA, as it targets autoantibody-producing plasma cells. Here, we present the first reported case of daratumumab used in conjunction with an erythropoiesisstimulating agent (ESA) to salvage refractory wAIHA in a patient with AIDS and bone marrow suppression.
Case Presentation
A middle aged man with HIV (undetectable viral load on antiretroviral treatment but CD4 persistently < 200, requiring chronic antimicrobial prophylaxis) was diagnosed with classic wAIHA in late 2021. The disease initially responded to corticosteroids, but relapsed repeatedly and eventually required IVIG, rituximab, danazol, and three immunosuppressive agents, none of which induced remission. Hemolysis worsened by fall 2024, with hemoglobin 5-6 g/dL despite high-dose corticosteroids and IVIG. Bone marrow biopsy was unrevealing, and he underwent splenectomy. However, recovery was complicated by cutaneous nocardiosis, iron overload, liver injury, and continued hemolysis. Eventually, reticulocytosis also ceased, and hemoglobin declined to 4-5 g/dL. Due to failure of standard therapies and to minimize further immunosuppression, weekly daratumumab injections were initiated, with weekly darbepoetin injections added to aid in compensatory hematopoiesis. With this combination, hemolysis indices improved, reticulocytosis picked up, and hemoglobin increased to 8-9 g/dL. However, the patient continued to struggle with infections, and he succumbed to drug-resistant bacterial sepsis in spring 2025.
Discussion
The patient had very complicated chronic and acute comorbidities, and some simplification was required in order to provide this summary. However, we hope this case adds to the literature on daratumumab as an effective new agent in refractory wAIHA, and also present a novel duo of therapies for patients who may struggle with bone marrow suppression in addition to autoimmune hemolysis. To our knowledge, this is the first reported case of the combination used in this manner.
Conclusions
Daratumumab is an effective and less immunosuppressive alternative for the treatment of heavily pretreated refractory wAIHA. Its combined use with ESA in patients with inadequate reticulocytosis should be studied further to clarify the efficacy and safety in this setting.
Background
Warm autoimmune hemolytic anemia (wAIHA) is traditionally treated with immunosuppresimmunosuppression, and management of refractory disease is often a challenge. The anti-CD38 antibody daratumumab is emerging as a promising treatment for refractory wAIHA, as it targets autoantibody-producing plasma cells. Here, we present the first reported case of daratumumab used in conjunction with an erythropoiesisstimulating agent (ESA) to salvage refractory wAIHA in a patient with AIDS and bone marrow suppression.
Case Presentation
A middle aged man with HIV (undetectable viral load on antiretroviral treatment but CD4 persistently < 200, requiring chronic antimicrobial prophylaxis) was diagnosed with classic wAIHA in late 2021. The disease initially responded to corticosteroids, but relapsed repeatedly and eventually required IVIG, rituximab, danazol, and three immunosuppressive agents, none of which induced remission. Hemolysis worsened by fall 2024, with hemoglobin 5-6 g/dL despite high-dose corticosteroids and IVIG. Bone marrow biopsy was unrevealing, and he underwent splenectomy. However, recovery was complicated by cutaneous nocardiosis, iron overload, liver injury, and continued hemolysis. Eventually, reticulocytosis also ceased, and hemoglobin declined to 4-5 g/dL. Due to failure of standard therapies and to minimize further immunosuppression, weekly daratumumab injections were initiated, with weekly darbepoetin injections added to aid in compensatory hematopoiesis. With this combination, hemolysis indices improved, reticulocytosis picked up, and hemoglobin increased to 8-9 g/dL. However, the patient continued to struggle with infections, and he succumbed to drug-resistant bacterial sepsis in spring 2025.
Discussion
The patient had very complicated chronic and acute comorbidities, and some simplification was required in order to provide this summary. However, we hope this case adds to the literature on daratumumab as an effective new agent in refractory wAIHA, and also present a novel duo of therapies for patients who may struggle with bone marrow suppression in addition to autoimmune hemolysis. To our knowledge, this is the first reported case of the combination used in this manner.
Conclusions
Daratumumab is an effective and less immunosuppressive alternative for the treatment of heavily pretreated refractory wAIHA. Its combined use with ESA in patients with inadequate reticulocytosis should be studied further to clarify the efficacy and safety in this setting.
Background
Warm autoimmune hemolytic anemia (wAIHA) is traditionally treated with immunosuppresimmunosuppression, and management of refractory disease is often a challenge. The anti-CD38 antibody daratumumab is emerging as a promising treatment for refractory wAIHA, as it targets autoantibody-producing plasma cells. Here, we present the first reported case of daratumumab used in conjunction with an erythropoiesisstimulating agent (ESA) to salvage refractory wAIHA in a patient with AIDS and bone marrow suppression.
Case Presentation
A middle aged man with HIV (undetectable viral load on antiretroviral treatment but CD4 persistently < 200, requiring chronic antimicrobial prophylaxis) was diagnosed with classic wAIHA in late 2021. The disease initially responded to corticosteroids, but relapsed repeatedly and eventually required IVIG, rituximab, danazol, and three immunosuppressive agents, none of which induced remission. Hemolysis worsened by fall 2024, with hemoglobin 5-6 g/dL despite high-dose corticosteroids and IVIG. Bone marrow biopsy was unrevealing, and he underwent splenectomy. However, recovery was complicated by cutaneous nocardiosis, iron overload, liver injury, and continued hemolysis. Eventually, reticulocytosis also ceased, and hemoglobin declined to 4-5 g/dL. Due to failure of standard therapies and to minimize further immunosuppression, weekly daratumumab injections were initiated, with weekly darbepoetin injections added to aid in compensatory hematopoiesis. With this combination, hemolysis indices improved, reticulocytosis picked up, and hemoglobin increased to 8-9 g/dL. However, the patient continued to struggle with infections, and he succumbed to drug-resistant bacterial sepsis in spring 2025.
Discussion
The patient had very complicated chronic and acute comorbidities, and some simplification was required in order to provide this summary. However, we hope this case adds to the literature on daratumumab as an effective new agent in refractory wAIHA, and also present a novel duo of therapies for patients who may struggle with bone marrow suppression in addition to autoimmune hemolysis. To our knowledge, this is the first reported case of the combination used in this manner.
Conclusions
Daratumumab is an effective and less immunosuppressive alternative for the treatment of heavily pretreated refractory wAIHA. Its combined use with ESA in patients with inadequate reticulocytosis should be studied further to clarify the efficacy and safety in this setting.
Successful Targeted Therapy with Alectinib in ALK-Positive Metastatic Pancreatic Cancer
Background
Pancreatic cancer has one of the highest mortality rates due to its typical late-stage diagnosis and subsequent limited surgical options. However, recent advances in molecular profiling offer hope for targeted therapies. We present a case of locally advanced pancreatic adenocarcinoma which progressed despite surgery and chemotherapy yet showed a positive respond to Alectinib.
Case Description
A 79-year-old male with medical history of tobacco use and ulcerative colitis presented to the clinic with 15lb unintentional weight loss over the past few months in 04/2021. Computed tomography (CT) showed dilated common bile duct due to 2.2 x 1.9 x 1.7 cm mass with no metastatic disease. Biopsy was consistent with pancreatic adenocarcinoma and patient completed 6 cycles of dose-reduced neoadjuvant gemcitabine and paclitaxel in late 2021 due to his severe neuropathy and ECOG. Subsequent CT and PET-CT showed stable disease prior to undergoing pylorus-sparing pancreatoduodenectomy and cholecystectomy with portal vein resection in 05/2022 with surgical pathology grading yPT4N2cM0. The follow- up PET scan in 09/2022 revealed new pulmonary and liver metastases, along with increased uptake in the pancreatic region, suggesting recurrent disease. Next generation sequencing (NGS) identified an ELM4-ALK chromosomal rearrangement on the surgical pathology. Given the patient’s cancer progression and concerns about chemotherapy tolerance, Alectinib, a second-generation ALK inhibitor more commonly used in lung cancer, was considered as a treatment option. Patient began Alectinib 10/2022 with no significant side effects and PET scan on 03/2023 and 06/2023 showing resolution of his lung nodules and liver lesions. Patient remained on Alectinib until he transitioned to hospice after an ischemic stroke in 03/2024.
Discussion
Pancreatic cancer urgently requires novel therapies as about 25% of patients harbor actionable molecular alterations that have led to the success of targeted therapies. ALK fusion genes are identified in multiple cancers, but the prevalence is only 0.16% in pancreatic ductal adenocarcinoma. Alectinib provided an extended progression free survival compared with standard chemotherapy in our patient. ALK inhibitors may demonstrate a remarkable response in metastatic pancreatic cancer even in poor candidates for standard chemotherapy highlighting the emphasis of NGS and targeted therapy options for pancreatic cancer to improve survival.
Background
Pancreatic cancer has one of the highest mortality rates due to its typical late-stage diagnosis and subsequent limited surgical options. However, recent advances in molecular profiling offer hope for targeted therapies. We present a case of locally advanced pancreatic adenocarcinoma which progressed despite surgery and chemotherapy yet showed a positive respond to Alectinib.
Case Description
A 79-year-old male with medical history of tobacco use and ulcerative colitis presented to the clinic with 15lb unintentional weight loss over the past few months in 04/2021. Computed tomography (CT) showed dilated common bile duct due to 2.2 x 1.9 x 1.7 cm mass with no metastatic disease. Biopsy was consistent with pancreatic adenocarcinoma and patient completed 6 cycles of dose-reduced neoadjuvant gemcitabine and paclitaxel in late 2021 due to his severe neuropathy and ECOG. Subsequent CT and PET-CT showed stable disease prior to undergoing pylorus-sparing pancreatoduodenectomy and cholecystectomy with portal vein resection in 05/2022 with surgical pathology grading yPT4N2cM0. The follow- up PET scan in 09/2022 revealed new pulmonary and liver metastases, along with increased uptake in the pancreatic region, suggesting recurrent disease. Next generation sequencing (NGS) identified an ELM4-ALK chromosomal rearrangement on the surgical pathology. Given the patient’s cancer progression and concerns about chemotherapy tolerance, Alectinib, a second-generation ALK inhibitor more commonly used in lung cancer, was considered as a treatment option. Patient began Alectinib 10/2022 with no significant side effects and PET scan on 03/2023 and 06/2023 showing resolution of his lung nodules and liver lesions. Patient remained on Alectinib until he transitioned to hospice after an ischemic stroke in 03/2024.
Discussion
Pancreatic cancer urgently requires novel therapies as about 25% of patients harbor actionable molecular alterations that have led to the success of targeted therapies. ALK fusion genes are identified in multiple cancers, but the prevalence is only 0.16% in pancreatic ductal adenocarcinoma. Alectinib provided an extended progression free survival compared with standard chemotherapy in our patient. ALK inhibitors may demonstrate a remarkable response in metastatic pancreatic cancer even in poor candidates for standard chemotherapy highlighting the emphasis of NGS and targeted therapy options for pancreatic cancer to improve survival.
Background
Pancreatic cancer has one of the highest mortality rates due to its typical late-stage diagnosis and subsequent limited surgical options. However, recent advances in molecular profiling offer hope for targeted therapies. We present a case of locally advanced pancreatic adenocarcinoma which progressed despite surgery and chemotherapy yet showed a positive respond to Alectinib.
Case Description
A 79-year-old male with medical history of tobacco use and ulcerative colitis presented to the clinic with 15lb unintentional weight loss over the past few months in 04/2021. Computed tomography (CT) showed dilated common bile duct due to 2.2 x 1.9 x 1.7 cm mass with no metastatic disease. Biopsy was consistent with pancreatic adenocarcinoma and patient completed 6 cycles of dose-reduced neoadjuvant gemcitabine and paclitaxel in late 2021 due to his severe neuropathy and ECOG. Subsequent CT and PET-CT showed stable disease prior to undergoing pylorus-sparing pancreatoduodenectomy and cholecystectomy with portal vein resection in 05/2022 with surgical pathology grading yPT4N2cM0. The follow- up PET scan in 09/2022 revealed new pulmonary and liver metastases, along with increased uptake in the pancreatic region, suggesting recurrent disease. Next generation sequencing (NGS) identified an ELM4-ALK chromosomal rearrangement on the surgical pathology. Given the patient’s cancer progression and concerns about chemotherapy tolerance, Alectinib, a second-generation ALK inhibitor more commonly used in lung cancer, was considered as a treatment option. Patient began Alectinib 10/2022 with no significant side effects and PET scan on 03/2023 and 06/2023 showing resolution of his lung nodules and liver lesions. Patient remained on Alectinib until he transitioned to hospice after an ischemic stroke in 03/2024.
Discussion
Pancreatic cancer urgently requires novel therapies as about 25% of patients harbor actionable molecular alterations that have led to the success of targeted therapies. ALK fusion genes are identified in multiple cancers, but the prevalence is only 0.16% in pancreatic ductal adenocarcinoma. Alectinib provided an extended progression free survival compared with standard chemotherapy in our patient. ALK inhibitors may demonstrate a remarkable response in metastatic pancreatic cancer even in poor candidates for standard chemotherapy highlighting the emphasis of NGS and targeted therapy options for pancreatic cancer to improve survival.